Exploratory analysis
Last updated: 2022-03-09
Checks: 7 0
Knit directory: RainDrop_biodiversity/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210406) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 73c5b98. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/site_libs/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/exploratory_analysis.Rmd) and HTML (docs/exploratory_analysis.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 73c5b98 | jjackson-eco | 2022-03-09 | wflow_publish(all = TRUE) |
| html | 73c5b98 | jjackson-eco | 2022-03-09 | wflow_publish(all = TRUE) |
| html | f105a1e | jjackson-eco | 2022-03-09 | Build site. |
| Rmd | 81990d5 | jjackson-eco | 2022-03-09 | wflow_publish(files = c(“analysis/exploratory_analysis.Rmd”, |
| html | 4bdd4a2 | jjackson-eco | 2021-06-15 | wflow_publish(files = c(“docs/index.html”, “docs/exploratory_analysis.html”, |
| html | 5b44884 | jjackson-eco | 2021-06-15 | Build site. |
| Rmd | cbc95ff | jjackson-eco | 2021-06-15 | wflow_publish(c(“.Rprofile”, “analysis/exploratory_analysis.Rmd”, |
| html | 0129bef | jjackson-eco | 2021-04-09 | Build site. |
| Rmd | 6e78a68 | jjackson-eco | 2021-04-09 | results update |
| html | ec6a27c | jjackson-eco | 2021-04-09 | Build site. |
| Rmd | 1e1c114 | jjackson-eco | 2021-04-09 | Adding species diversity |
| html | cc963a5 | jjackson-eco | 2021-04-08 | Build site. |
| Rmd | b427f16 | jjackson-eco | 2021-04-08 | exploration update 2 |
| html | 104d3f2 | jjackson-eco | 2021-04-08 | Build site. |
| Rmd | 0faa75d | jjackson-eco | 2021-04-08 | exploration update |
| html | c69c14a | jjackson-eco | 2021-04-08 | Build site. |
| Rmd | f8308af | jjackson-eco | 2021-04-08 | exploration for biomass |
| html | 7a899b4 | jjackson-eco | 2021-04-07 | Build site. |
| Rmd | 9381cc9 | jjackson-eco | 2021-04-07 | wflow_publish(files = c(“README.md”, "analysis/_site.yml“,”analysis/exploratory_analysis.Rmd", |
Here we present some exploratory analysis and plots investigating general patterns in the two biodiversity measures estimated at the RainDrop site as part of the Drought-Net experiment, above-ground biomass (biomass) and species diversity (percent_cover).
First some housekeeping. We’ll start by loading the necessary packages for exploring the data, the data itself and then specifying the colour palette for the treatments.
# packages
library(tidyverse)
library(patchwork)
library(vegan)
# load data
load("../../RainDropRobotics/Data/raindrop_biodiversity_2016_2021.RData",
verbose = TRUE)Loading objects:
biomass
percent_cover# colours
raindrop_colours <-
tibble(treatment = c("Ambient", "Control", "Drought", "Irrigated"),
num = rep(100,4),
colour = c("#61D94E", "#BFBFBF", "#EB8344", "#6ECCFF"))
ggplot(raindrop_colours, aes(x = treatment, y = num, fill = treatment)) +
geom_col() +
geom_text(aes(label = colour, x = treatment, y = 50), size = 3) +
scale_fill_manual(values = raindrop_colours$colour) +
theme_void()
1. Above-ground biomass
Data summary
Here, cuttings of 1m x 0.25m strips (from ~1cm above the ground) from each treatment and block are sorted by functional group and after drying at 70\(^\circ\)C for over 48 hours their dry mass is weighed in grams. Measurements occur twice per year- one harvest in June in the middle of the growing season and one and in September at the end of the growing season.
Here a summary of the data:
glimpse(biomass)Rows: 1,273
Columns: 6
$ year <dbl> 2016, 2016, 2016, 2016, 2016, 2016, 2016, 2016, 2016, 2016, ~
$ harvest <chr> "Mid", "Mid", "Mid", "Mid", "Mid", "Mid", "Mid", "Mid", "Mid~
$ block <chr> "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", "A", ~
$ treatment <chr> "Ambient", "Ambient", "Ambient", "Ambient", "Ambient", "Ambi~
$ group <chr> "Bryophytes", "Dead", "Forbs", "Graminoids", "Legumes", "Woo~
$ biomass_g <dbl> 0.7, 4.5, 16.5, 49.7, 7.7, 0.0, 0.3, 2.4, 20.0, 46.7, 3.6, 1~- $year - year of measurement (integer 2016-2021)
- $harvest - point of the growing season when harvest occur (category “Mid” and “End”)
- $block - block of experiment (category A-E)
- $treatment - experimental treatment for Drought-Net (category “Ambient”, “Control”, “Drought” and “Irrigated”)
- $group - functional group of measurement (category “Bryophytes”, “Dead”, “Forbs”, “Graminoids”, “Legumes” and “Woody”)
- $biomass_g - above-ground dry mass in grams (continuous) per 0.2 meter squared
Biomass variable
Let us first have a look at the response variable biomass_g. We’ll remove any zero observations of biomass (Need to know if 0 observations are true zeros or procedural) and convert to a standarised format (grams per meter squared). If we plot out the frequency histogram of the raw data, we can see there is some strong positive skew in this variable, because of the differences in biomass scale between our functional groups (i.e. grasses very over-represented).
biomass <- biomass %>%
filter(biomass_g > 0) %>%
mutate(biomass_g = biomass_g * 4)
ggplot(biomass, aes(x = biomass_g)) +
geom_histogram(bins = 20) +
labs(x = expression(paste("Above-ground biomass (g/", m^2, ")")), y = "Frequency") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank())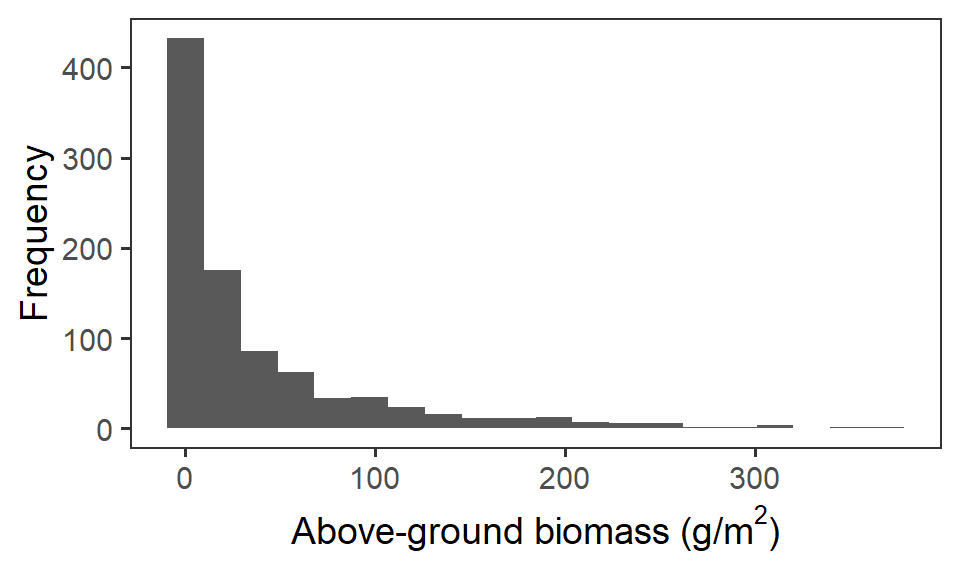
So, instead, for statistical analysis and visualisation, we take the natural logarithm \(\ln\) of the raw biomass value.
biomass <- biomass %>%
mutate(biomass_log = log(biomass_g))
ggplot(biomass, aes(x = biomass_log)) +
geom_histogram(bins = 20) +
labs(x = expression(paste(ln," Above-ground biomass")), y = "Frequency") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank())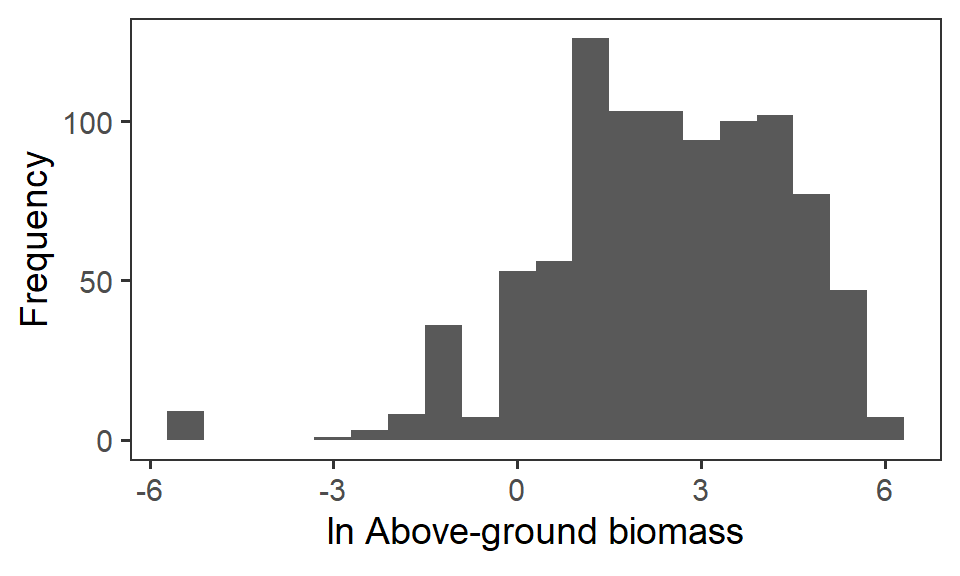
Using the ln biomass variable, we can explore patterns relating to our drought treatments, functional groups, temporal patterns and any potential block effects.
Total biomass
The first interesting question to explore is how biomass overall, which indicates primary productivity, is affected by the rainfall treatments. Here we need to sum up the biomass across functional groups. We again look at the \(\ln\) total biomass too. We present here the density plots (violins) with median (total biomass) and mean (\(\ln\) total biomass) points (large holes) as well as underlying raw data (small points).
totbiomass <- biomass %>%
group_by(year, harvest, block, treatment) %>%
summarise(tot_biomass = sum(biomass_g),
log_tot_biomass = log(tot_biomass + 1)) %>% # avoiding 0 biomasses
ungroup()And now we can look at how this is affected by the rainfall treatments.
tb_1 <- totbiomass %>%
ggplot(aes(x = treatment, y = tot_biomass,
fill = treatment)) +
geom_violin() +
stat_summary(fun = median, geom = "point",
size = 6, shape = 21, fill = "white") +
geom_jitter(width = 0.1, alpha = 0.4, size = 1) +
scale_fill_manual(values = raindrop_colours$colour,
guide = F) +
labs(x = "Rainfall treatment", y = expression(paste("Above-ground biomass (g/", m^2, ")"))) +
theme_bw(base_size = 20) +
theme(panel.grid = element_blank())
tb_2 <- totbiomass %>%
ggplot(aes(x = treatment, y = log_tot_biomass,
fill = treatment)) +
geom_violin() +
stat_summary(fun = mean, geom = "point",
size = 6, shape = 21, fill = "white") +
geom_jitter(width = 0.1, alpha = 0.4, size = 1)+
scale_fill_manual(values = raindrop_colours$colour,
guide = F) +
labs(x = "Rainfall treatment",
y = expression(paste(ln, " Total above-ground biomass"))) +
theme_bw(base_size = 20) +
theme(panel.grid = element_blank())
tb_1 + tb_2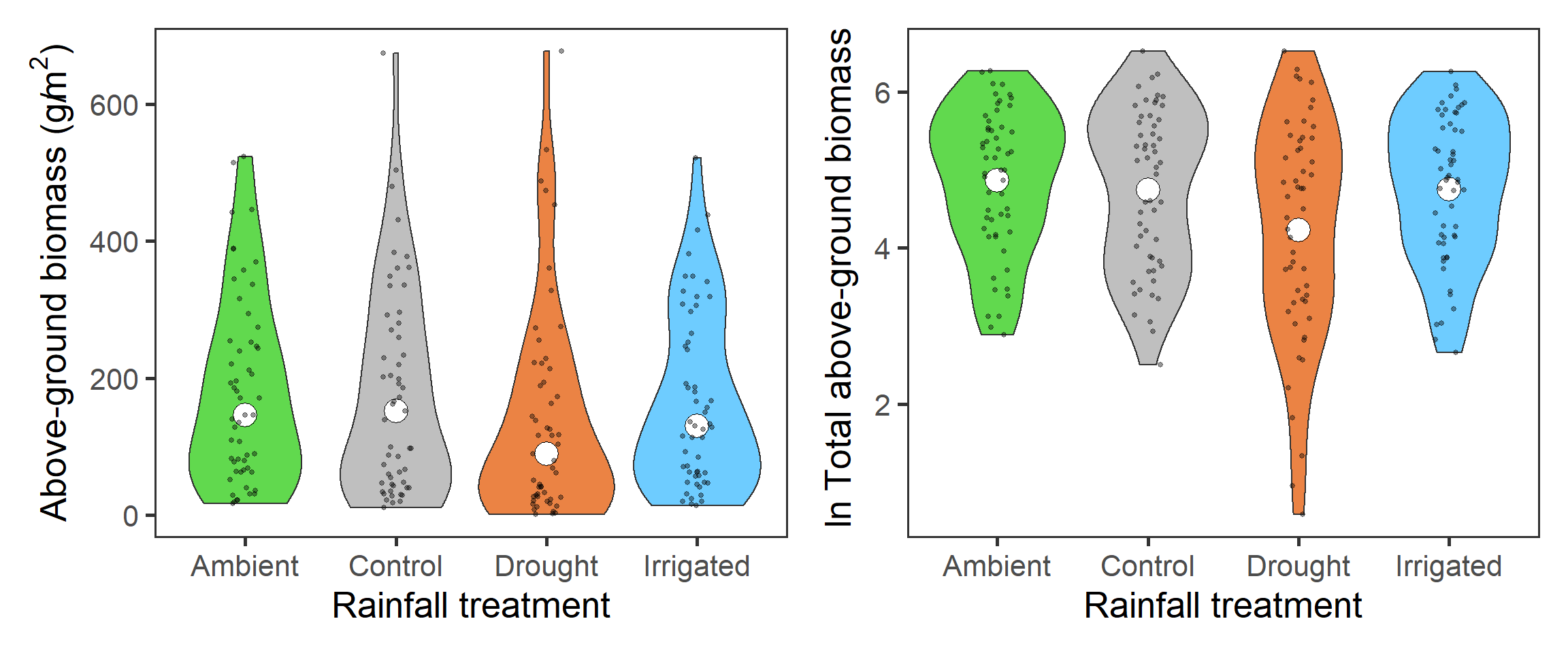
Across all functional groups, years and blocks, it looks like there is a reduction in total biomass associated with the drought treatment.
Now we’ll look at potential temporal effects i.e. whether there is a change in total biomass over time. We first have to add in temporal information to the year and harvest columns, and then we can plot out the total biomass.
# adding temporal information
totbiomass <- totbiomass %>%
mutate(month = if_else(harvest == "End", 9, 6),
date = as.Date(paste0(year,"-",month,"-15")),
harvest = factor(harvest, levels = c("Mid", "End")))
# plots
totbiomass %>%
ggplot(aes(x = date, y = log_tot_biomass, colour = treatment,
group = treatment, shape = harvest)) +
stat_summary(geom = "line", fun = mean, size = 0.9) +
stat_summary(geom = "point", fun = mean, size = 3) +
scale_colour_manual(values = raindrop_colours$colour) +
labs(x = "Year", y = expression(paste("Mean ", ln, " total biomass")),
colour = "Treatment", shape = "Harvest") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank())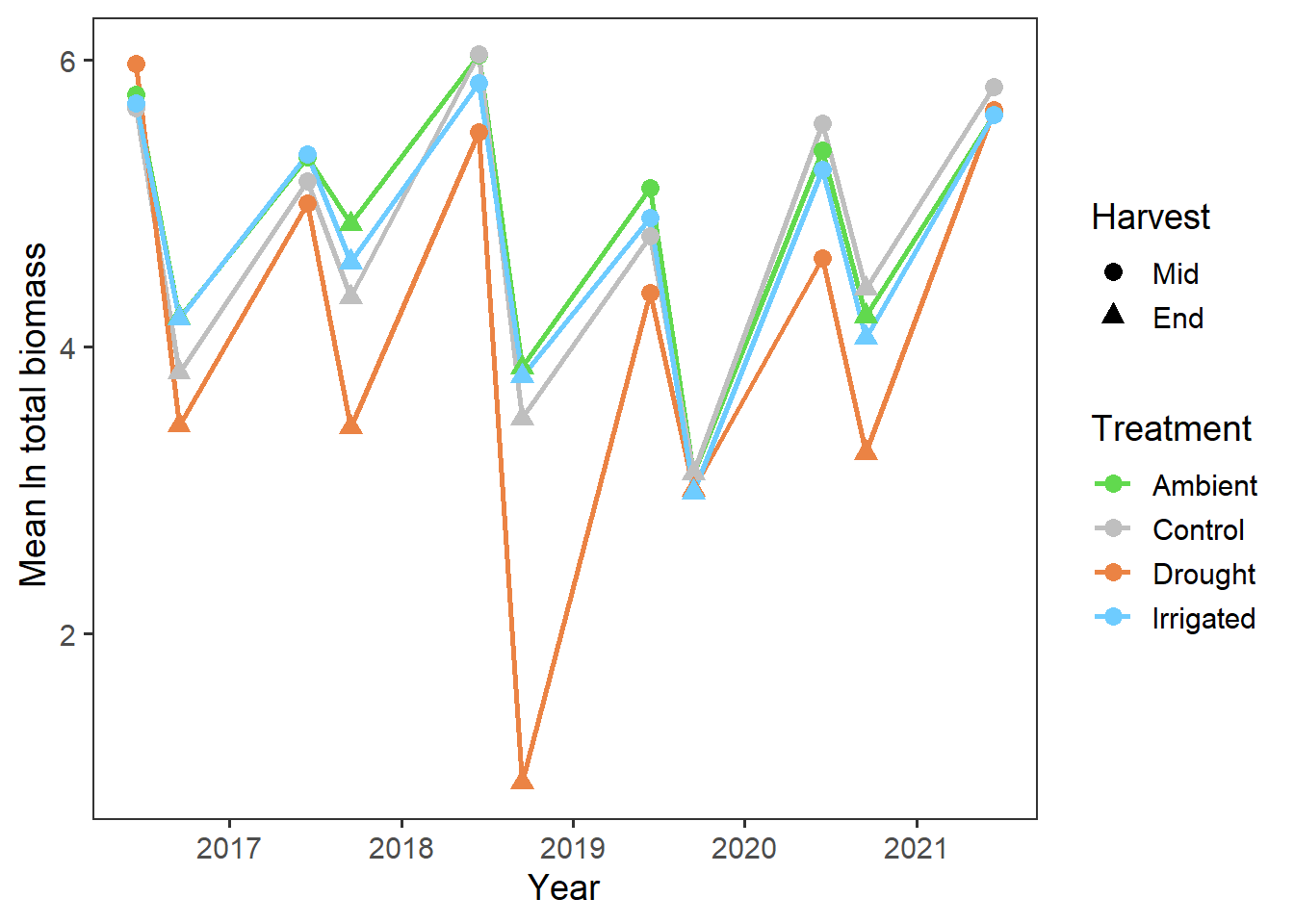
And also splitting out the harvests, as the end of the growing season has lower biomass
totbiomass %>%
ggplot(aes(x = year, y = log_tot_biomass, colour = treatment,
group = treatment)) +
stat_summary(geom = "line", fun = mean, size = 0.9) +
stat_summary(geom = "point", fun = mean, size = 3) +
scale_colour_manual(values = raindrop_colours$colour) +
labs(x = "Year", y = expression(paste("Mean ", ln, " total biomass")),
colour = "Treatment") +
facet_wrap(~harvest) +
theme_bw(base_size = 20) +
theme(panel.grid = element_blank(),
strip.background = element_blank())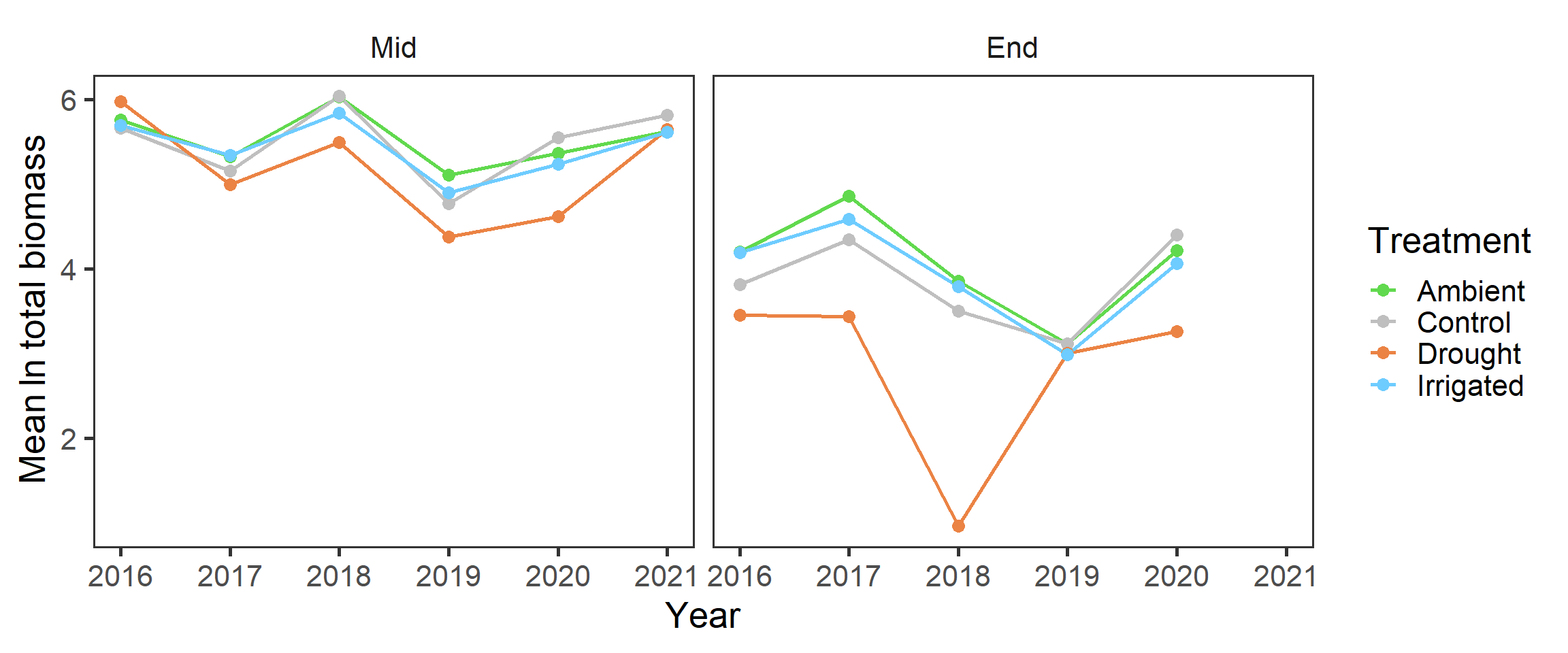
It looks like there may have been declines in total biomass over the years (perhaps reflecting wider climatic patterns?), but this also seems to be more prominent in the drought treatment.
Finally for total biomass, we want to look if we have any evidence for block effects at the experimental site. Here we will repeat the first treatment violin plot, but splitting by block. Here each violin is a different block (A-E from left to right) with the 50% quantile.
totbiomass %>%
ggplot(aes(x = treatment, y = log_tot_biomass,
fill = treatment,
group = interaction(treatment, block))) +
geom_violin(draw_quantiles = 0.5) +
scale_fill_manual(values = raindrop_colours$colour,
guide = F) +
labs(x = "Rainfall treatment",
y = expression(paste(ln, " Total above-ground biomass"))) +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank())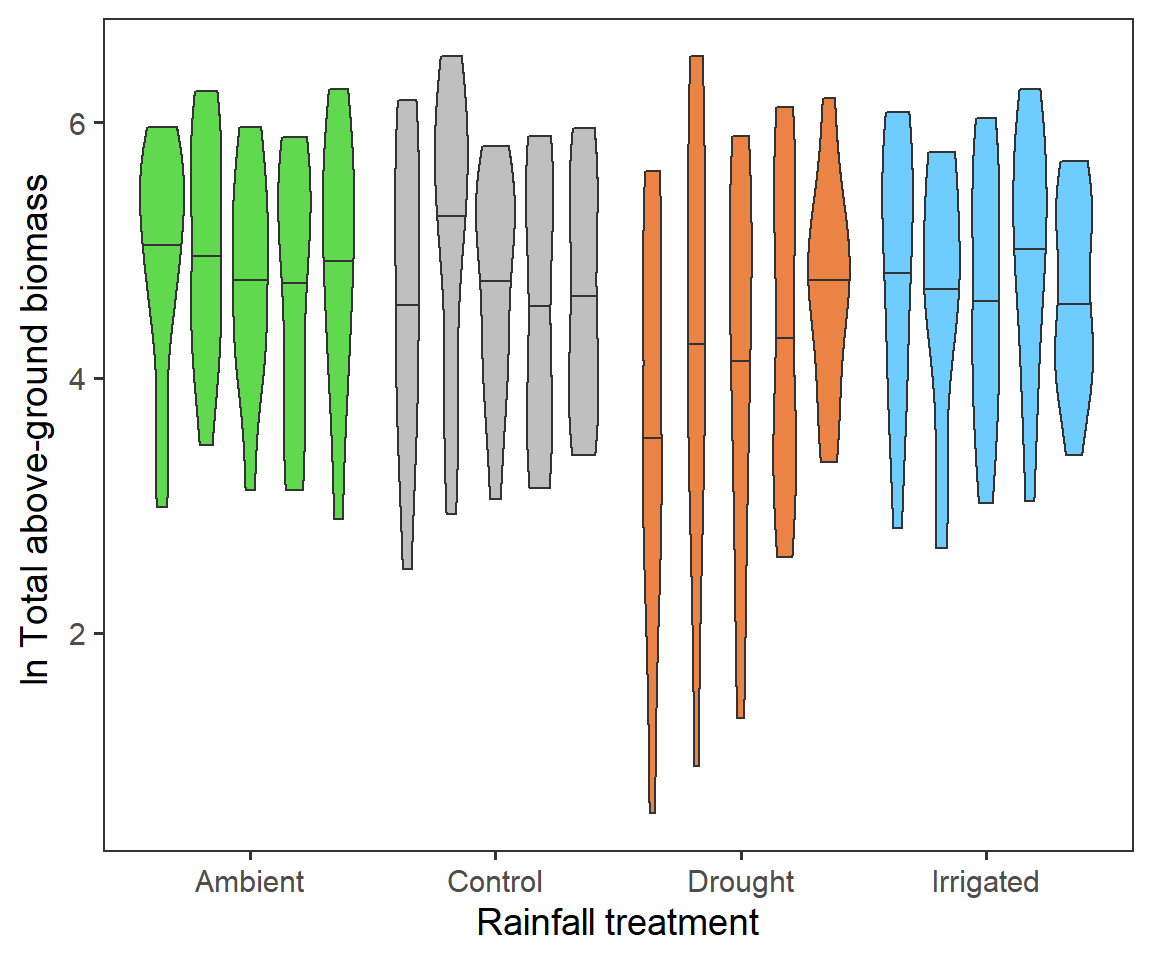
It seems that largely blocks are consistent across the RainDrop site.
Group-level biomass
Now we can go a little further and look at whether we observe any differences in biomass between different functional groups, which occur at different proportions. First, lets look at the overall differences in biomass, or relative biomass, between functional groups.
biomass %>%
filter(is.na(group) == FALSE) %>%
ggplot(aes(x = group, y = biomass_log,
fill = group)) +
geom_violin() +
stat_summary(fun = mean, geom = "point",
size = 5, shape = 21, fill = "white") +
geom_jitter(width = 0.15, alpha = 0.2, size = 1) +
scale_fill_brewer(palette = "Set2", guide = F) +
labs(x = "Functional group",
y = expression(paste(ln, " Above-ground biomass"))) +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank())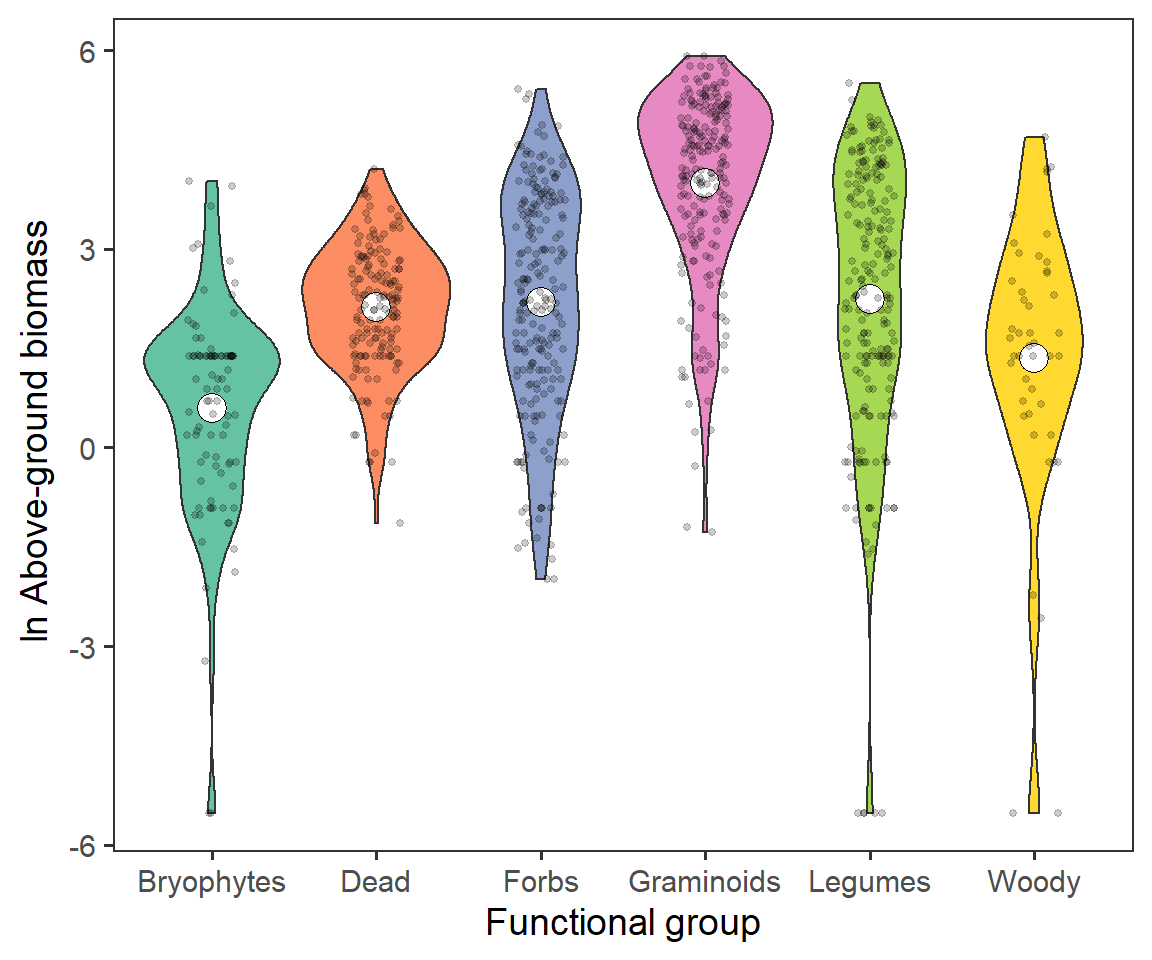
We can see that there is a large representation (as expected) of Graminoids, and lower representation of Woody species and Bryophytes.
Now lets look at the rainfall treatments by functional group.
biomass %>%
filter(is.na(group) == FALSE) %>%
ggplot(aes(x = treatment, y = biomass_log,
fill = treatment)) +
geom_violin() +
stat_summary(fun = mean, geom = "point",
size = 5, shape = 21, fill = "white") +
geom_jitter(width = 0.15, alpha = 0.2, size = 1) +
scale_fill_manual(values = raindrop_colours$colour,
guide = F) +
labs(x = "Rainfall treatment",
y = expression(paste(ln, " Above-ground biomass"))) +
facet_wrap(~ group, ncol = 2, scales ="free") +
theme_bw(base_size = 22) +
theme(panel.grid = element_blank(),
strip.background = element_blank())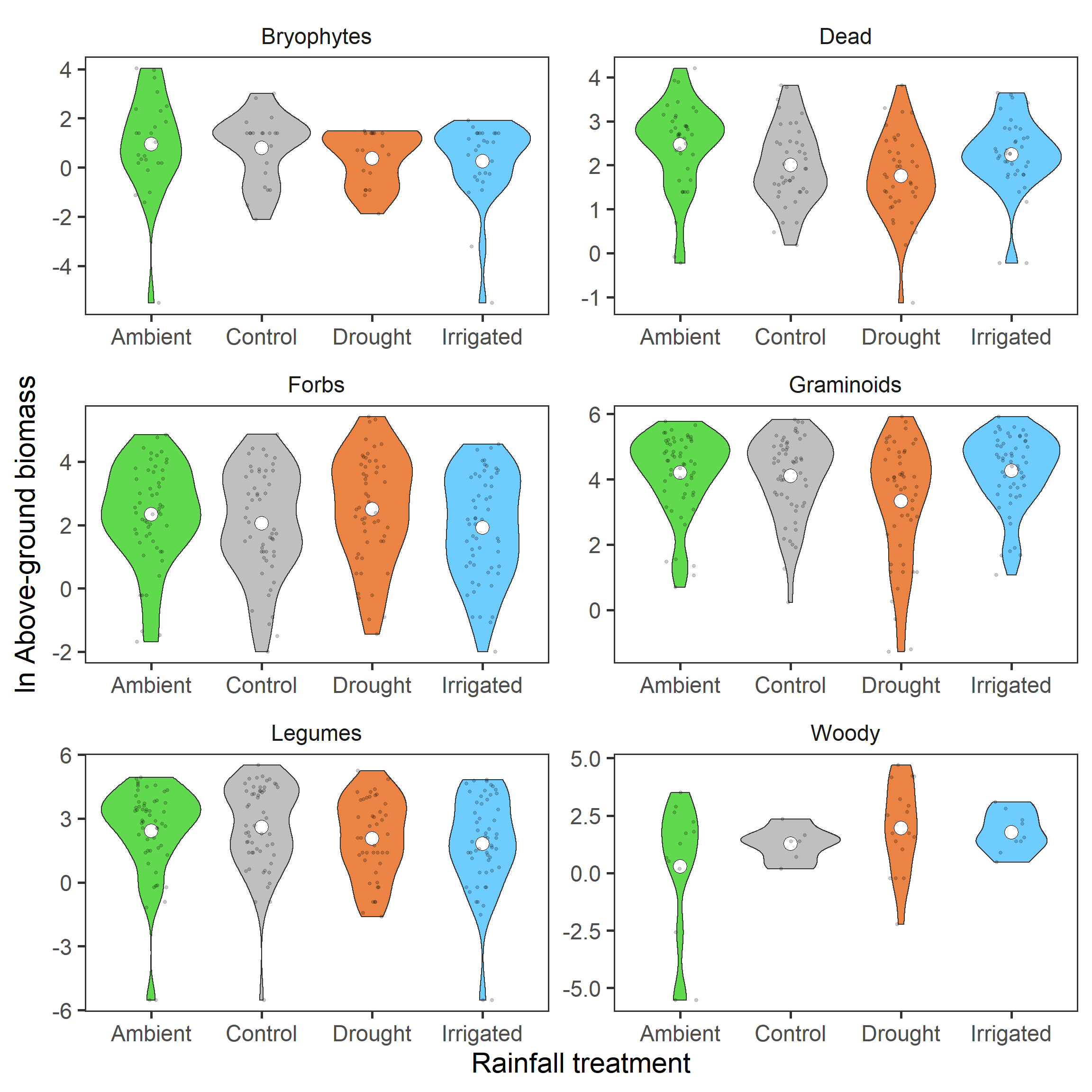
It isn’t as clear hear as total biomass. However, for the group with the most representation, the Graminoids, this reduction in the drought treatment looks to be maintained.
Finally we can look at whether there are differences in the temporal patterns between the functional groups.
# adding temporal information
biomass <- biomass %>%
filter(is.na(group) == FALSE) %>%
mutate(month = if_else(harvest == "End", 9, 6),
date = as.Date(paste0(year,"-",month,"-15")),
harvest = factor(harvest, levels = c("Mid", "End")))
# plot
biomass %>%
ggplot(aes(x = date, y = biomass_log, colour = treatment,
group = treatment, shape = harvest)) +
stat_summary(geom = "line", fun = mean, size = 0.9) +
stat_summary(geom = "point", fun = mean, size = 3) +
scale_colour_manual(values = raindrop_colours$colour) +
facet_wrap(~group) +
labs(x = "Year", y = expression(paste(ln, " Above-ground biomass")),
colour = "Treatment", shape = "Harvest") +
theme_bw(base_size = 22) +
theme(panel.grid = element_blank(),
strip.background = element_blank())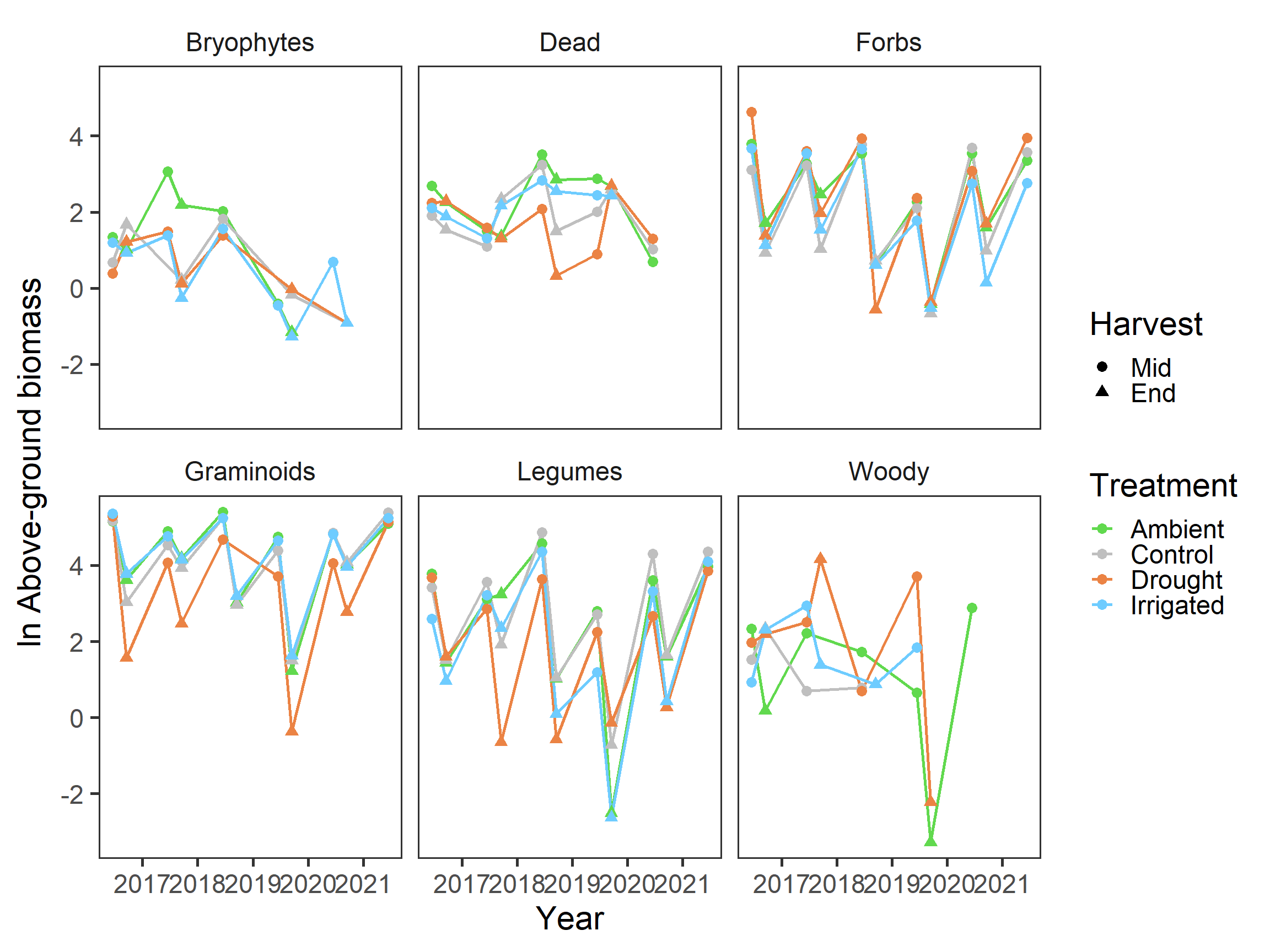
Again, specific patterns are hard to discern here, but it looks like biomass is decreasing slightly again in general.
2. Species diversity
Data summary
The species diversity data is in in the form of percentage cover of individually id’ species in a 1m x 1m quadrant in the center of each experimental plot. These percentage cover measurements were generally made at the peak of the growing season (June), but in 2016 there were recordings in both June and September.
Here a summary of the data:
glimpse(percent_cover)Rows: 3,531
Columns: 8
$ year <dbl> 2018, 2016, 2016, 2016, 2016, 2016, 2016, 2016, 2016, 20~
$ month <chr> "June", "June", "June", "June", "June", "June", "June", ~
$ block <chr> "C", "B", "B", "C", "E", "A", "E", "C", "D", "B", "D", "~
$ treatment <chr> "Control", "Ambient", "Control", "Irrigated", "Control",~
$ species_level <chr> "No", "Yes", "Yes", "Yes", "Yes", "Yes", "Yes", "Yes", "~
$ species <chr> "Acer_sp", "Agrimonia_eupatoria", "Agrimonia_eupatoria",~
$ group <chr> NA, "Forb", "Forb", "Forb", "Forb", "Forb", "Forb", "For~
$ percent_cover <dbl> 1.0, 1.0, 2.0, 4.0, 4.0, 5.0, 5.0, 7.0, 8.0, 10.0, 15.0,~- $year - year of measurement (integer 2016-2021)
- $month - month of measurement (category “June” and “Sept”)
- $block - block of experiment (category A-E)
- $treatment - experimental treatment for Drought-Net (category “Ambient”, “Control”, “Drought” and “Irrigated”)
- $species_level - Was the measurement made at the level of species? (category “Yes”, “No”)
- $species - Binomial name of species recorded in the measurement (category, 138 unique records)
- $group - Functional group/plant type of the species recorded (category “Forb”, “Grass”, “Woody”, “Legume”)
- $percent_cover - percentage cover of the species in a 1m x 1m quadrat
For the purpose of analysis here, we are interested in species diversity, and so we only want to include observations for which we have species-level data. Also, we want to clean any NA observations.
percent_cover <- percent_cover %>%
drop_na(percent_cover) %>%
filter(species_level == "Yes")Diversity Indices
Our first key question regarding the percentage cover is how the rainfall treatments are influencing the community diversity of the plots. For this we will calculate some key biodiversity indices:
Species richness \(R = \text{number of types (i.e. species)}\)
Simpson’s index \(\lambda = \sum_{i = 1}^{R} p_i^2\)
Shannon-Wiener index \(H' = - \sum_{i = 1}^{R}p_i\ln p_i\)
where \(p_i\) is the relative proportion of type (here species) \(i\).
Here we calculate these diversity indices for each plot (i.e. each year, month, block, treatment combination)
diversity <- percent_cover %>%
# first convert percentages to a proportion
mutate(proportion = percent_cover/100) %>%
group_by(year, month, block, treatment) %>%
# diversity indices for each group
summarise(richness = n(),
simpsons = sum(proportion^2),
shannon = -sum(proportion * log(proportion))) %>%
ungroup() %>%
pivot_longer(cols = c(richness, simpsons, shannon),
names_to = "index") %>%
mutate(index_lab = case_when(
index == "richness" ~ "Species richness",
index == "simpsons" ~ "Simpson's index",
index == "shannon" ~ "Shannon-Wiener index"
))Diversity indices exploratory plots
Now we can explore how the diversity indices for each plot are related to the experimental treatments, temporal patterns, and any block effects.
First the treatment effects
diversity %>%
ggplot(aes(x = treatment, y = value,
fill = treatment)) +
geom_violin() +
stat_summary(fun = mean, geom = "point",
size = 6, shape = 21, fill = "white") +
geom_jitter(width = 0.1, alpha = 0.4, size = 1) +
scale_fill_manual(values = raindrop_colours$colour,
guide = F) +
labs(x = "Rainfall treatment", y = "Species diverity index") +
facet_wrap(~index_lab, ncol = 1, scales = "free") +
theme_bw(base_size = 20) +
theme(panel.grid = element_blank(),
strip.background = element_blank())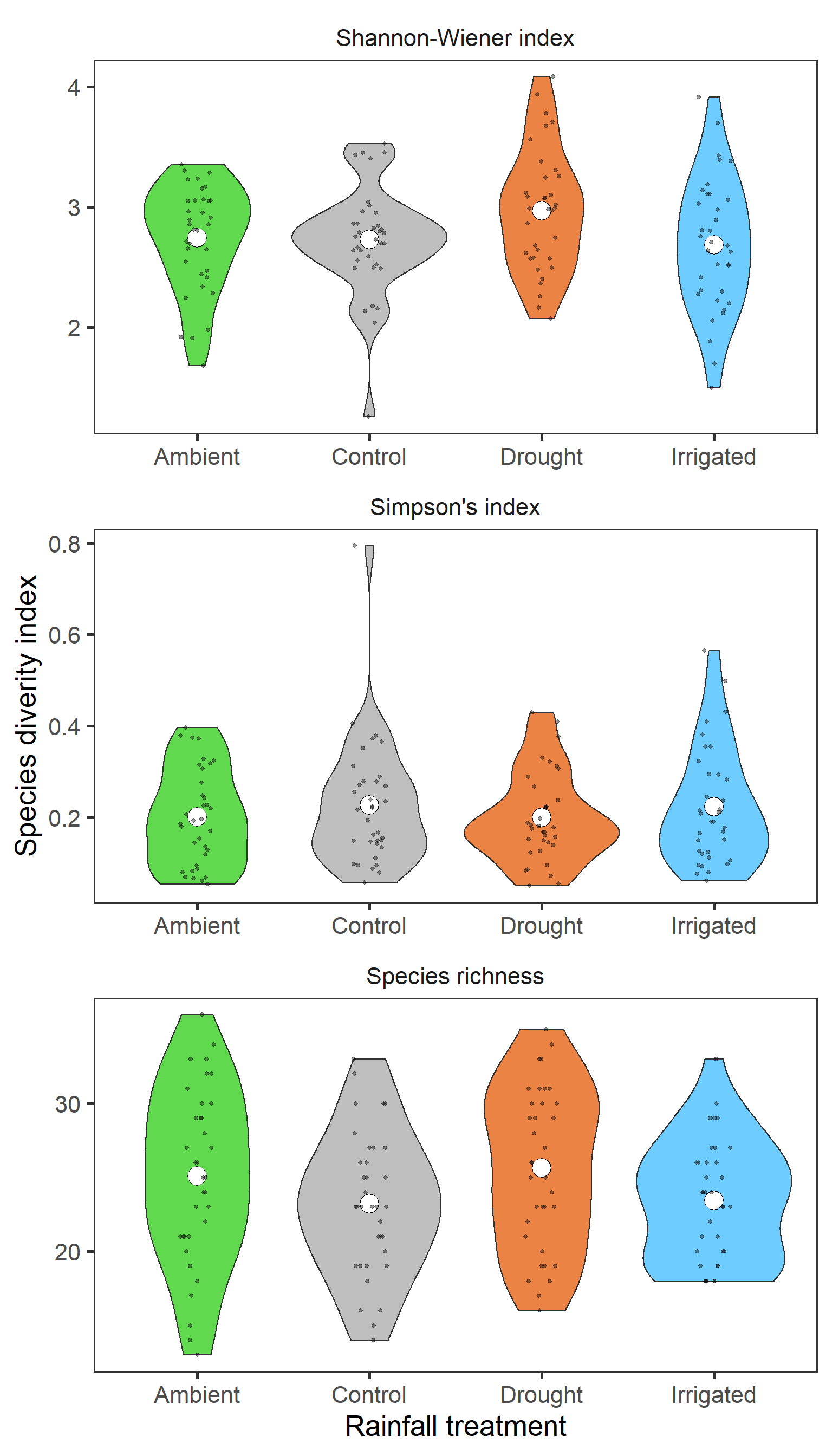
There isn’t a clear pattern here. But interestingly, it actually looks like for Shannon-Wiener, diversity is greatest in the drought treatment.
And now for potential temporal patterns
diversity %>%
mutate(month = if_else(month == "June", 6, 9),
date = as.Date(paste0(year,"-",month,"-15"))) %>%
ggplot(aes(x = date, y = value, colour = treatment)) +
stat_summary(geom = "line", fun = mean, size = 0.9) +
stat_summary(geom = "point", fun = mean, size = 3) +
scale_colour_manual(values = raindrop_colours$colour) +
labs(x = "Year", y = "Mean diversity index", colour = "Treatment") +
facet_wrap(~ index_lab, scales = "free", ncol = 1) +
theme_bw(base_size = 20) +
theme(panel.grid = element_blank(),
strip.background = element_blank())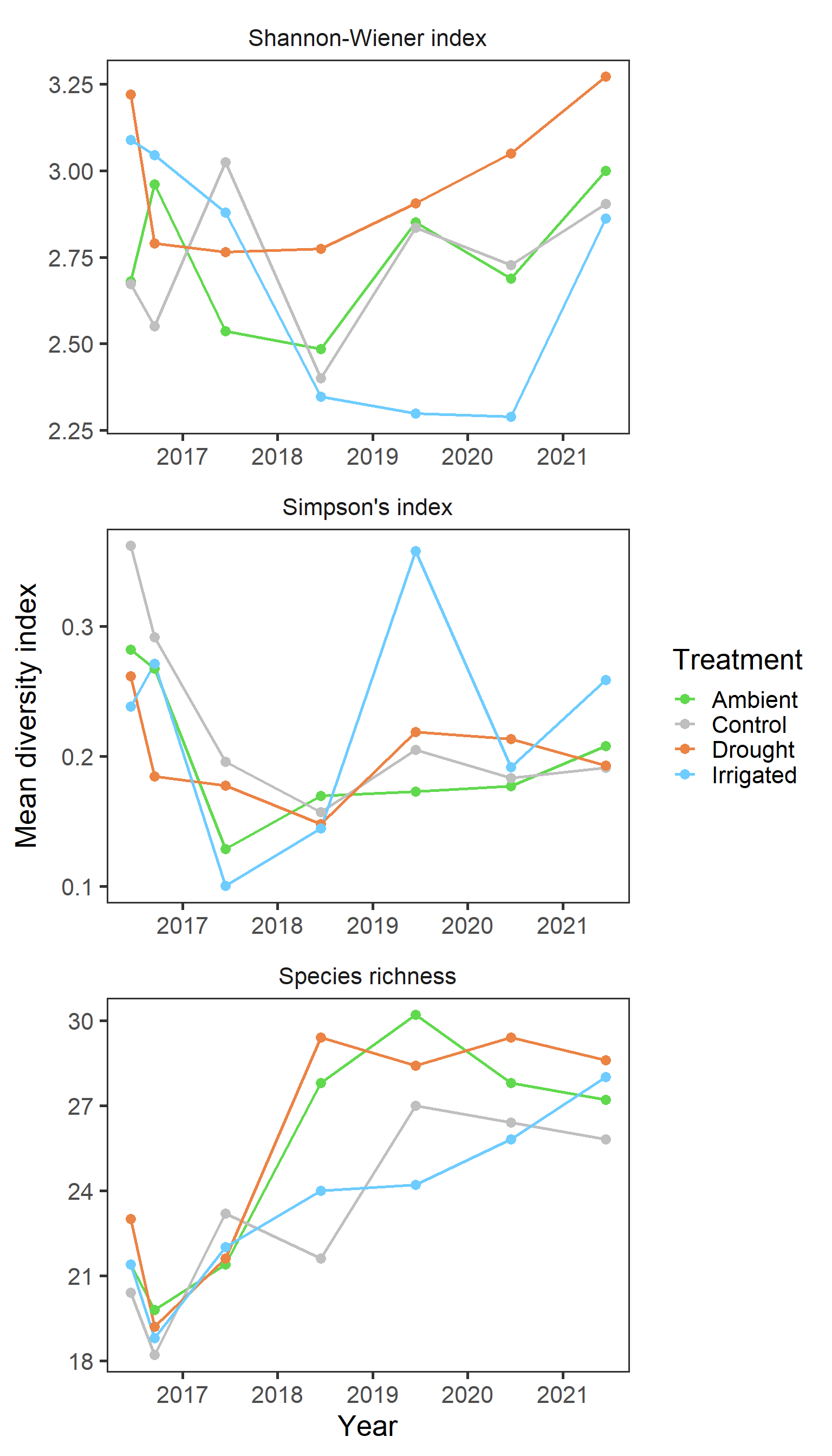
Again clear patterns are not sticking out with general indices. And finally we explore any potential block effects
diversity %>%
ggplot(aes(x = treatment, y = value,
fill = treatment,
group = interaction(treatment, block))) +
geom_violin(draw_quantiles = 0.5) +
scale_fill_manual(values = raindrop_colours$colour,
guide = F) +
labs(x = "Rainfall treatment",
y = "Species diversity index") +
facet_wrap(~ index_lab, scales = "free", ncol = 1) +
theme_bw(base_size = 20) +
theme(panel.grid = element_blank(),
strip.background = element_blank())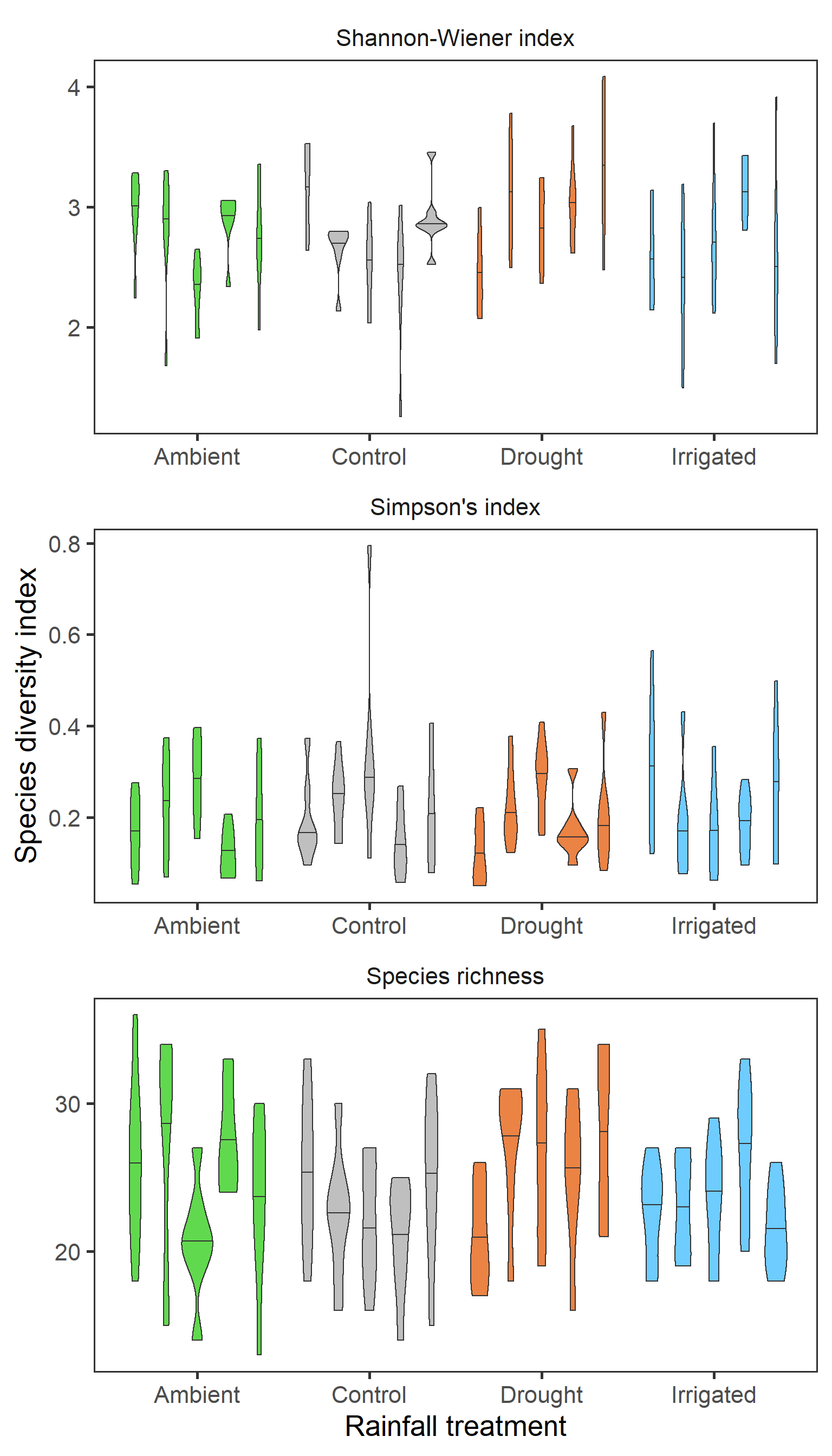
Functional group biodiversity
While there doesn’t seem to be a clear biodiversity signal overall, it is possible that something is occuring at the level of functional groups.
diversity_gr <- percent_cover %>%
# first convert percentages to a proportion
mutate(proportion = percent_cover/100) %>%
filter(group %in% c("Grass", "Forb", "Legume")) %>%
group_by(year, month, block, treatment, group) %>%
# diversity indices for each group
summarise(richness = n(),
simpsons = sum(proportion^2),
shannon = -sum(proportion * log(proportion))) %>%
ungroup() %>%
pivot_longer(cols = c(richness, simpsons, shannon),
names_to = "index") %>%
mutate(index_lab = case_when(
index == "richness" ~ "Species richness",
index == "simpsons" ~ "Simpson's index",
index == "shannon" ~ "Shannon-Wiener index"
))
diversity_gr %>%
filter(index_lab == "Species richness") %>%
ggplot(aes(x = treatment, y = value,
fill = treatment)) +
geom_violin() +
stat_summary(fun = mean, geom = "point",
size = 6, shape = 21, fill = "white") +
geom_jitter(width = 0.1, alpha = 0.4, size = 1) +
scale_fill_manual(values = raindrop_colours$colour,
guide = F) +
labs(x = "Rainfall treatment", y = "Species richness") +
facet_wrap(~ group, ncol = 3, scales = "free") +
theme_bw(base_size = 20) +
theme(panel.grid = element_blank(),
strip.background = element_blank())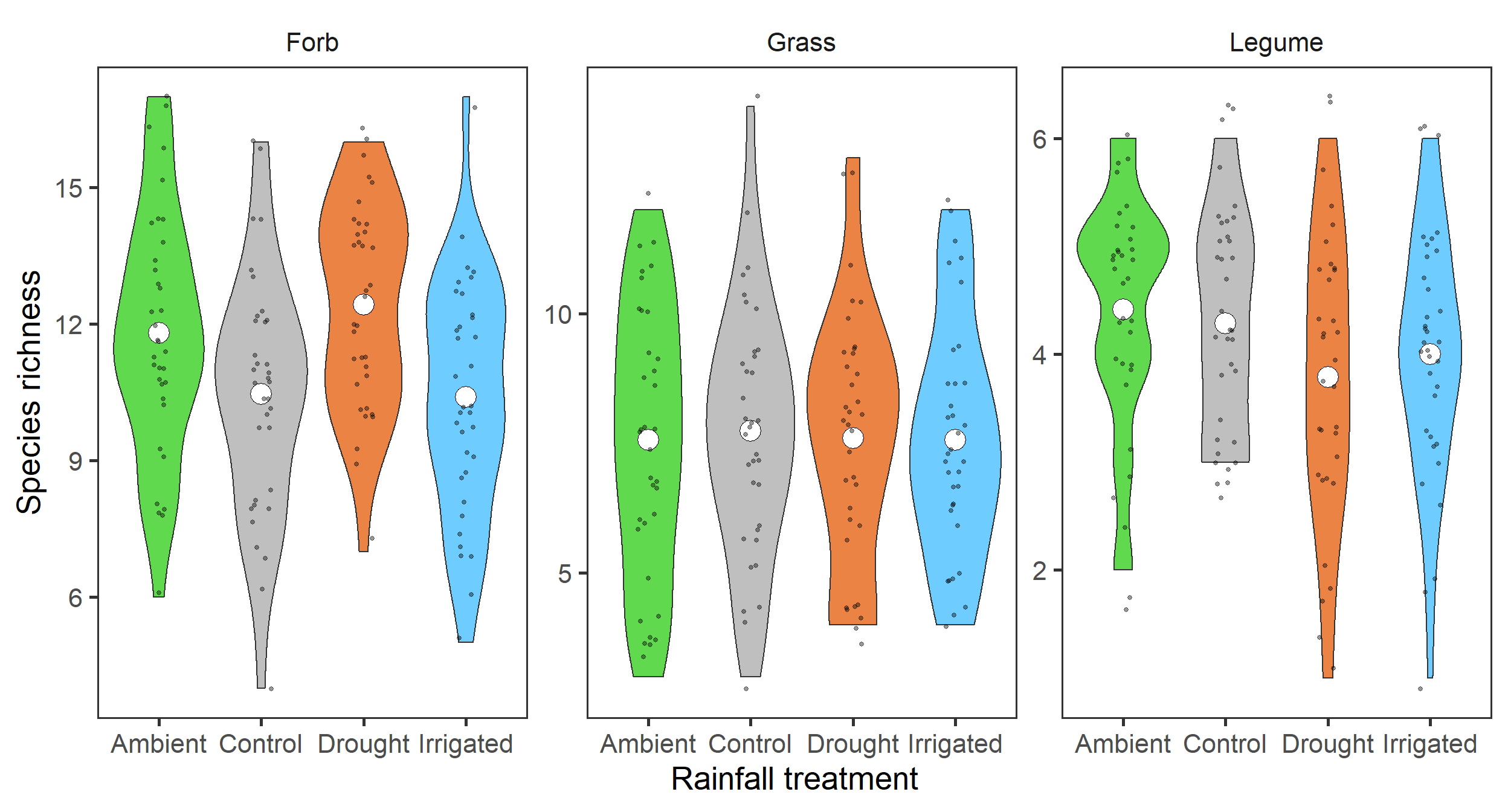
diversity_gr %>%
filter(index_lab == "Shannon-Wiener index") %>%
ggplot(aes(x = treatment, y = value,
fill = treatment)) +
geom_violin() +
stat_summary(fun = mean, geom = "point",
size = 6, shape = 21, fill = "white") +
geom_jitter(width = 0.1, alpha = 0.4, size = 1) +
scale_fill_manual(values = raindrop_colours$colour,
guide = F) +
labs(x = "Rainfall treatment", y = "Shannon-Wiener index") +
facet_wrap(~ group, ncol = 3, scales = "free") +
theme_bw(base_size = 20) +
theme(panel.grid = element_blank(),
strip.background = element_blank())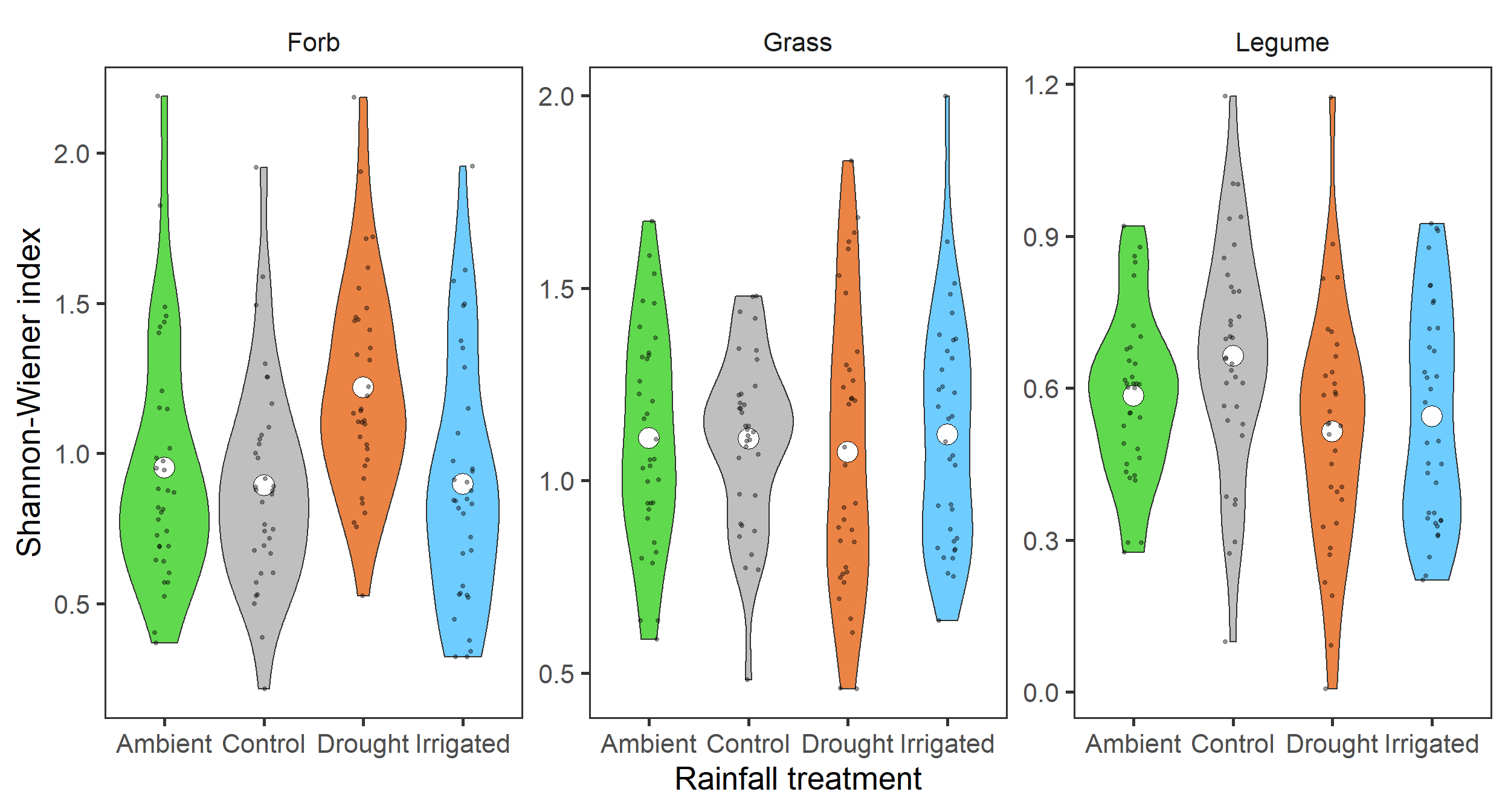
Here, it does look like there may be some weak effects. Specifically, it looks like there may be more forb species in the Drought treatment.
3. Community structure
While there may not be overall patterns in species diversity, what is interesting whether there are shifts to community structure. So, here we can perform a simple non-metric multidimensional scaling (NMDS) analysis, to look at changes to community structure with our treatments, general plant types, and also whether there are key species.
Here we perform the NMDS analysis using the vegan package, with two dimensions for 122 species in total and using the default bray distances. Firstly some cleaning though, to convert the percent cover data to a species by sample (our plots) matrix.
# data frame of the community matrix
species_community_df <- percent_cover %>%
mutate(sample = paste(month, year, block, treatment, sep = "_")) %>%
filter(species_level == "Yes") %>%
dplyr::select(sample, species, percent_cover) %>%
pivot_wider(id_cols = sample, names_from = species, values_from = percent_cover,
values_fill = 0)
# convert to a matrix
species_community_mat <- as.matrix(species_community_df[,2:125])
rownames(species_community_mat) <- species_community_df$sample
# have a look
glimpse(species_community_mat)
# the NMDS
raindrop_NMDS <- metaMDS(species_community_mat, k = 3, trymax = 1000) # Seems to be 3 dimensions with the new dataAnd now we can pull out the NMDS scores (3 dimensions), and explore their patterns in relation to the RainDrop study. First we look at the rainfall treatments in relation to community structure. Focusing on the first 2 dimensions for initial plotting.
# scores and species points
rd_scores <- as.data.frame(raindrop_NMDS$points)
rd_species <- as.data.frame(raindrop_NMDS$species)
# converting to a nice data frame
rd_scores <- rd_scores %>%
mutate(plot = rownames(.)) %>%
mutate(month = as.character(map(strsplit(plot, "_"), 1)),
year = as.numeric(map(strsplit(plot, "_"), 2)),
block = as.character(map(strsplit(plot, "_"), 3)),
treatment = as.character(map(strsplit(plot, "_"), 4)))
# and for species
species_types <- percent_cover %>%
group_by(species) %>%
summarise(group = group[1])
rd_species <- rd_species %>%
mutate(species = rownames(.)) %>%
left_join(x = ., y = species_types, by = "species")
# treatment plot
rd_scores %>%
ggplot(aes(x = MDS1, y = MDS2, colour = treatment)) +
geom_point(alpha = 0.8, size = 4) +
stat_ellipse(level = 0.8, size = 1, show.legend = F) +
scale_colour_manual(values = raindrop_colours$colour) +
labs(x = "NMDS1", y = "NMDS2", colour = "Rainfall\ntreatment") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank())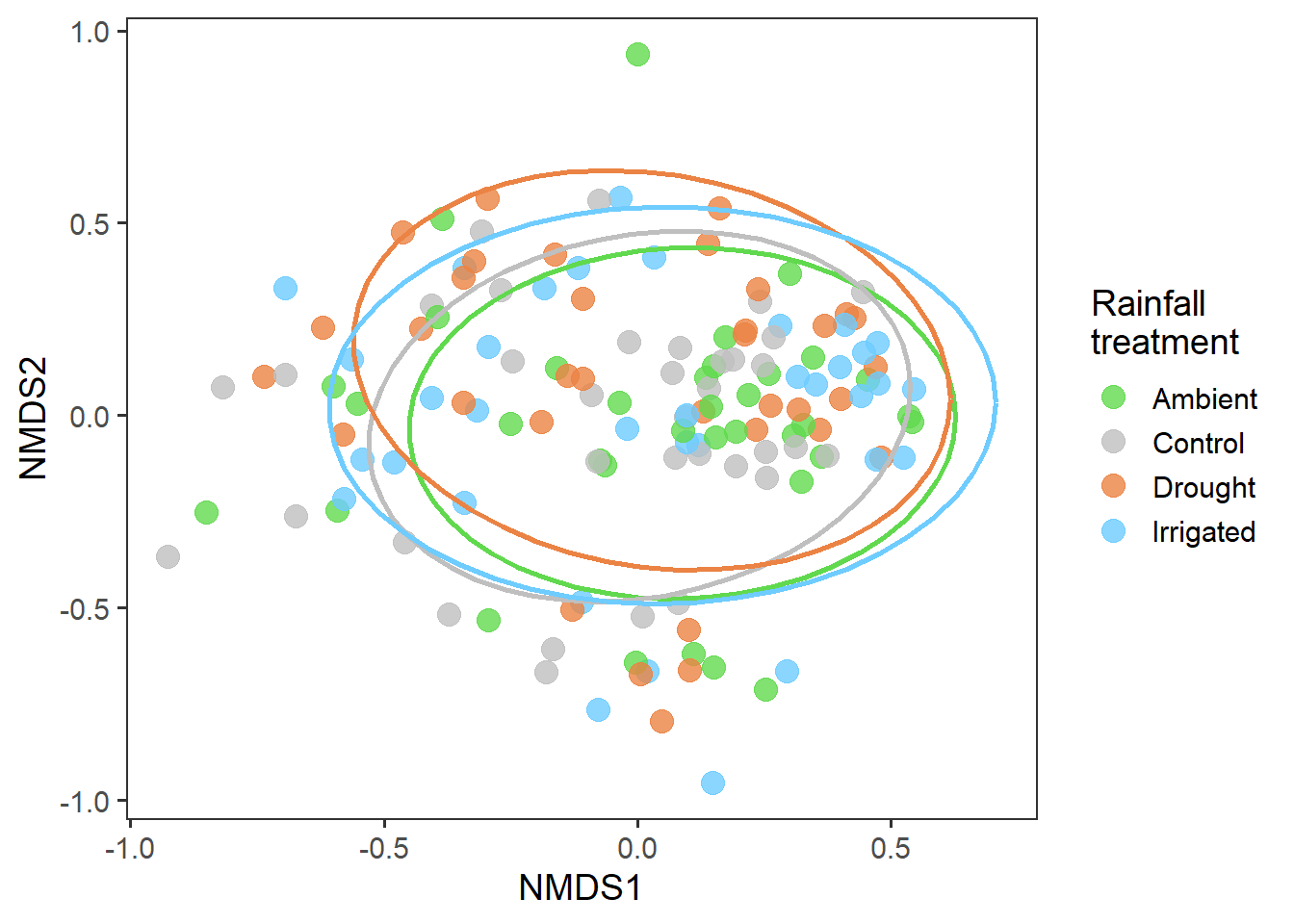
And now whether there are any interesting temporal effects
rd_scores %>%
ggplot(aes(x = MDS1, y = MDS2, colour = as.factor(year))) +
geom_point(alpha = 0.8, size = 4) +
stat_ellipse(level = 0.8, size = 1, show.legend = F) +
scale_colour_viridis_d() +
labs(x = "NMDS1", y = "NMDS2", colour = "Year") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank())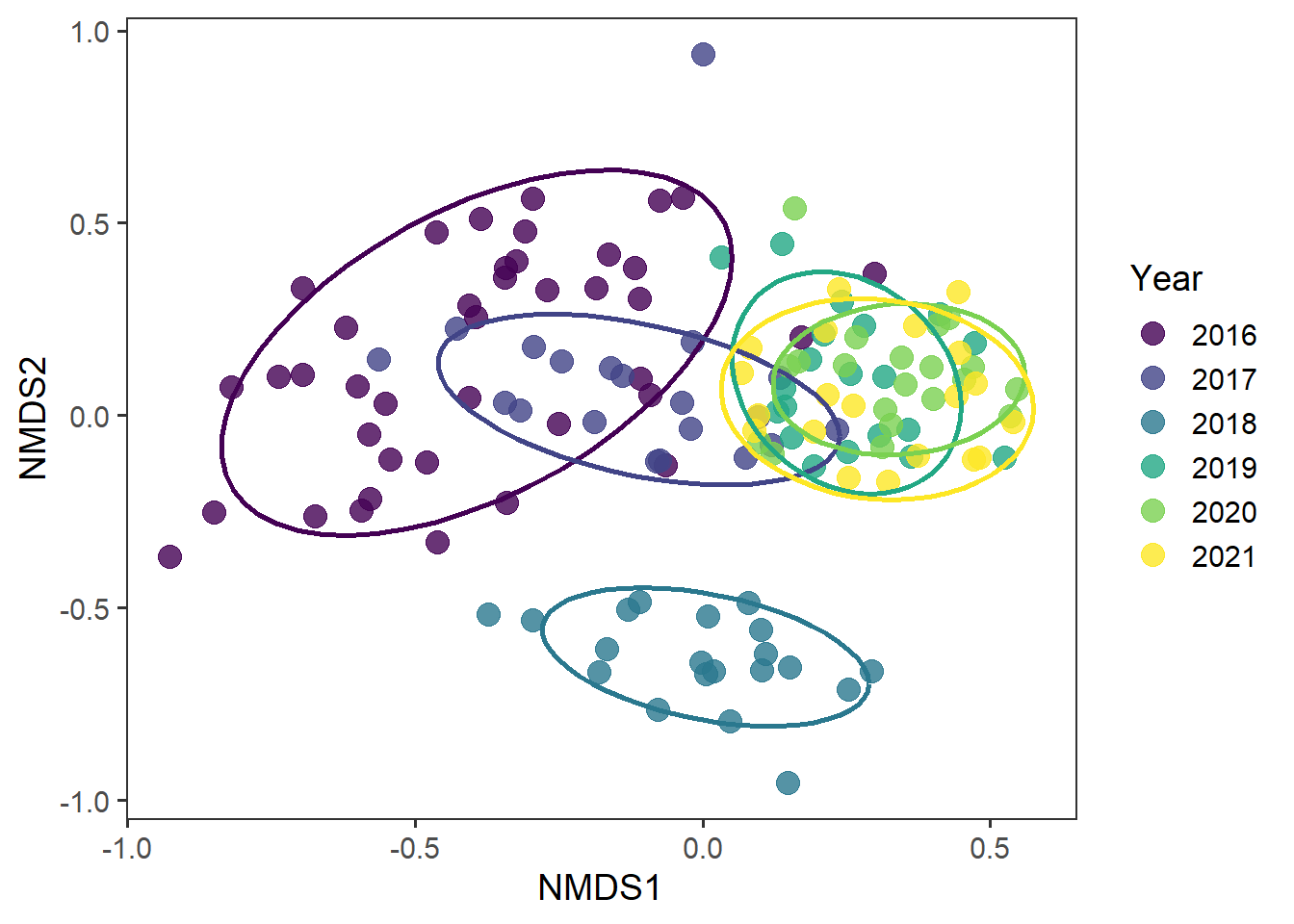
It does look like there are interesting temporal patterns in community structure here. Lets look at treatment by year all connected together.
Exploring quadrat-level trajectories
To dig a little deeper into these temporal effects, we can now explore the community trajectories through time for each observation quadrat. The overall data looks like this:
rd_scores %>%
mutate(block_treatment = paste0(block, "_", treatment)) %>%
ggplot(aes(x = MDS1, y = MDS2, colour = year, shape = treatment,
group = block_treatment)) +
geom_line(size = 1) +
geom_point(size = 4) +
labs(x = "NMDS1", y = "NMDS2", colour = "Year") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank())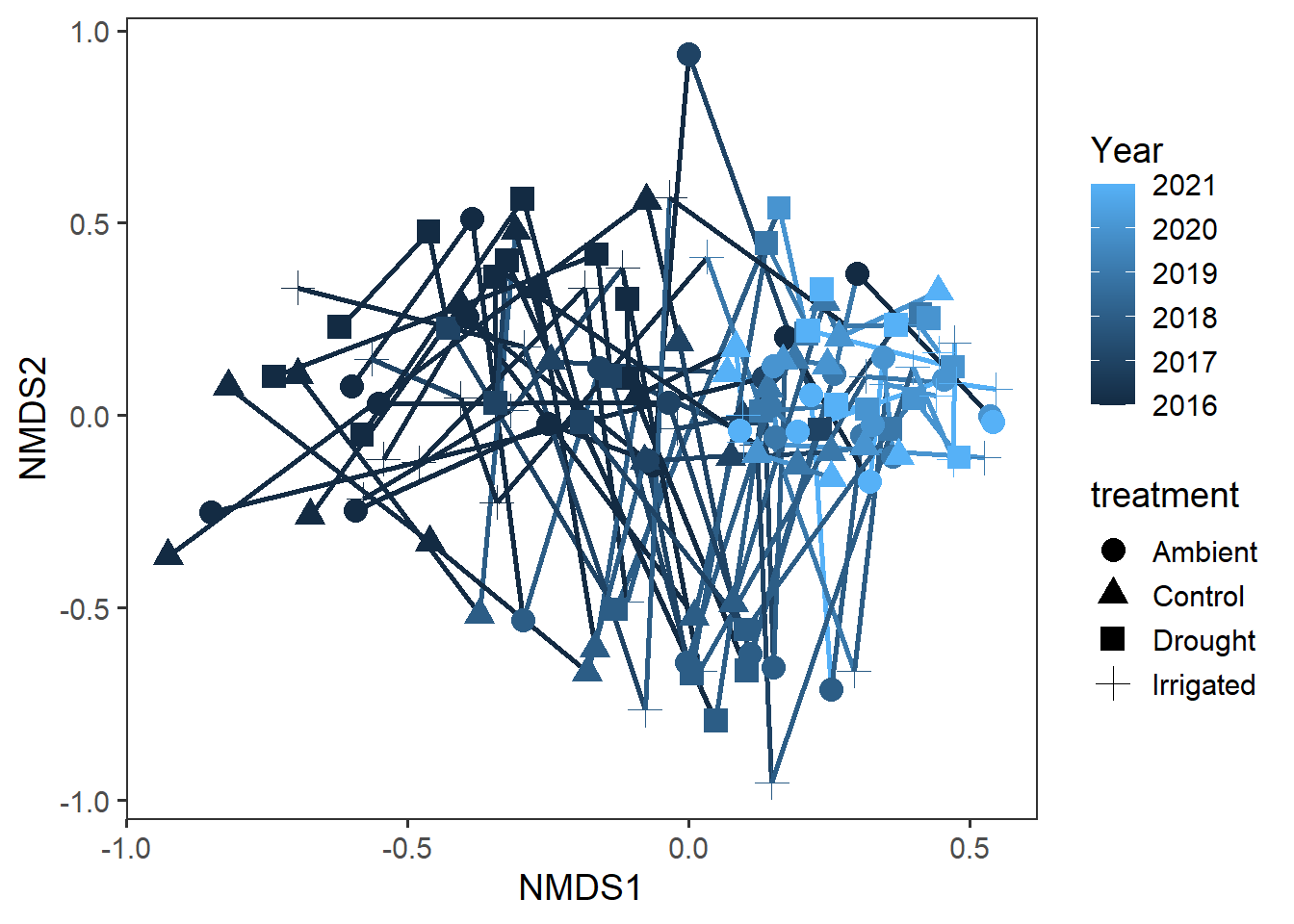
And now we will look at large plots exploring the trajectories of the three community structure components for each quadrat.
rd_scores %>%
mutate(block_treatment = paste0(block, "_", treatment)) %>%
ggplot(aes(x = year, y = MDS1, colour = treatment,
group = block_treatment)) +
geom_hline(yintercept = 0, size = 0.5) +
geom_line(size = 1) +
geom_point(size = 4) +
facet_wrap(~ treatment, ncol = 4) +
scale_colour_manual(values = raindrop_colours$colour, guide = "none") +
labs(x = "Year", y = "NMDS1", colour = "Treatment") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank(),
strip.background = element_blank())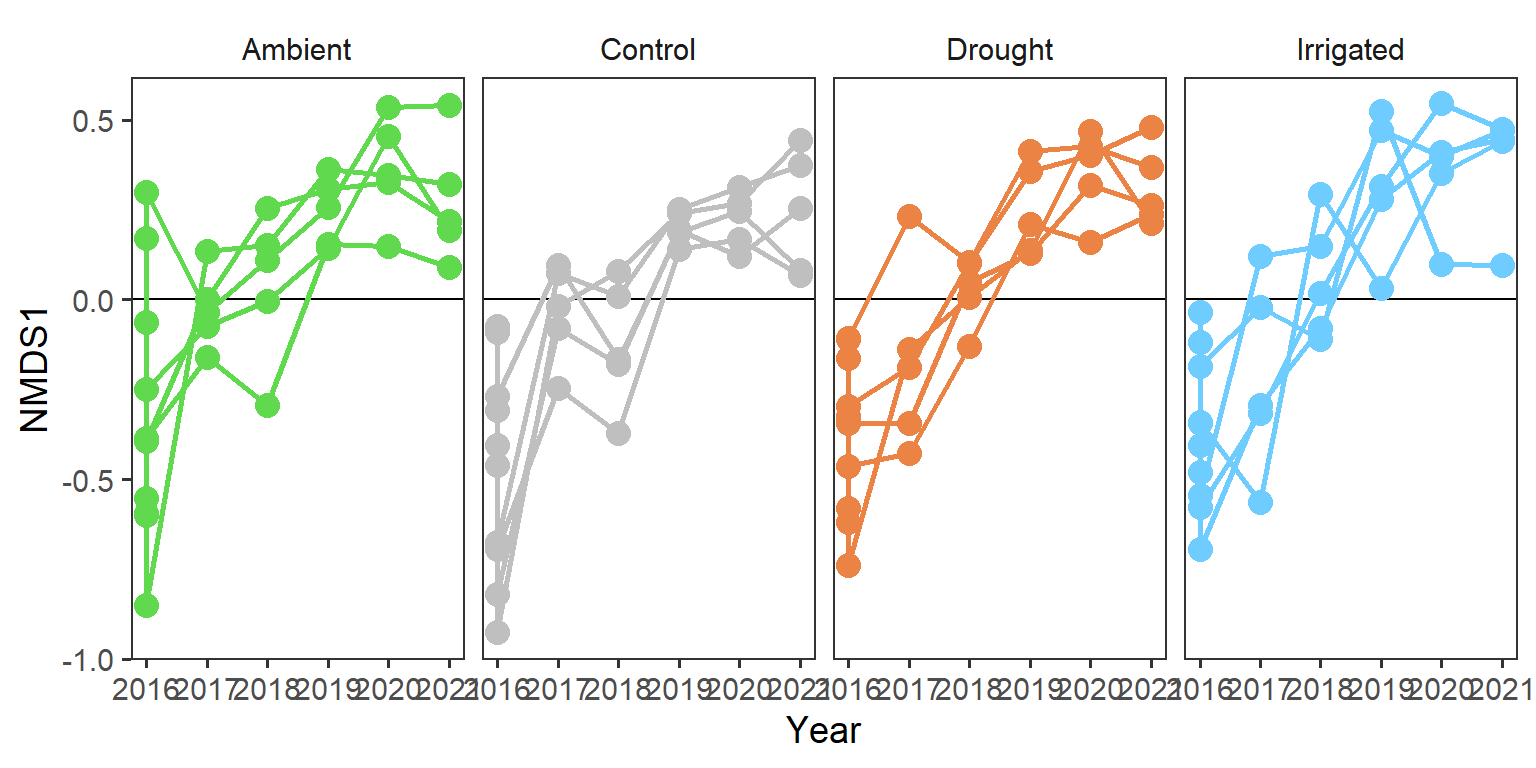
rd_scores %>%
mutate(block_treatment = paste0(block, "_", treatment)) %>%
ggplot(aes(x = year, y = MDS2, colour = treatment,
group = block_treatment)) +
geom_hline(yintercept = 0, size = 0.5) +
geom_line(size = 1) +
geom_point(size = 4) +
facet_wrap(~ treatment, ncol = 4) +
scale_colour_manual(values = raindrop_colours$colour, guide = "none") +
labs(x = "Year", y = "NMDS2", colour = "Treatment") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank(),
strip.background = element_blank())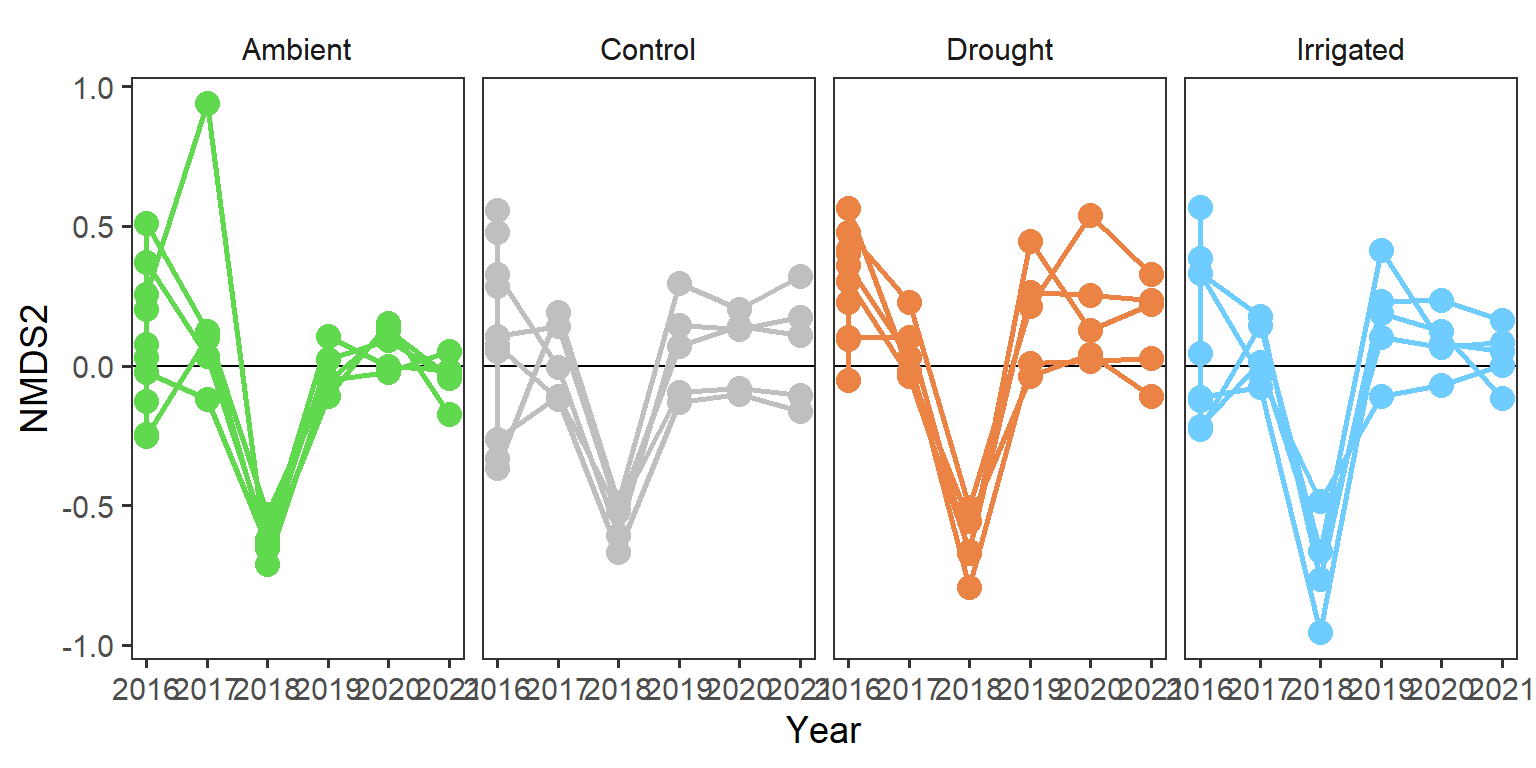
rd_scores %>%
mutate(block_treatment = paste0(block, "_", treatment)) %>%
ggplot(aes(x = year, y = MDS3, colour = treatment,
group = block_treatment)) +
geom_hline(yintercept = 0, size = 0.5) +
geom_line(size = 1) +
geom_point(size = 4) +
facet_wrap(~ treatment, ncol = 4) +
scale_colour_manual(values = raindrop_colours$colour, guide = "none") +
labs(x = "Year", y = "NMDS3", colour = "Treatment") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank(),
strip.background = element_blank())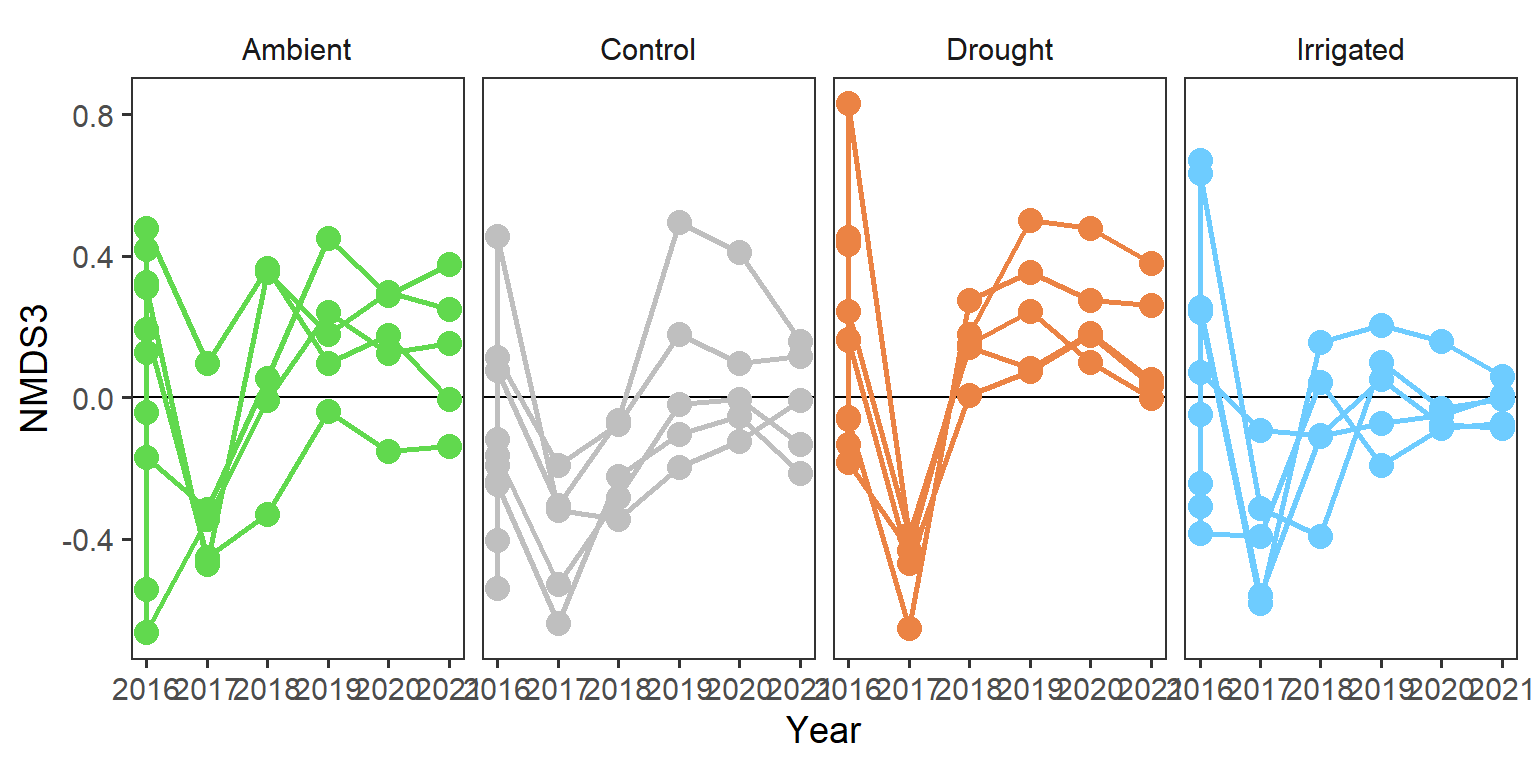
Species importance
And we can now see whether there are important groups of species or groups of species that may be driving community structure differences. This is essentially the ‘loading’ of each species to the two NMDS axes.
# pulling the 95% quantiles to identify most extreme data
q1 <- quantile(rd_species$MDS1, c(0.025, 0.975))
q2 <- quantile(rd_species$MDS2, c(0.025, 0.975))
rd_species_extreme <- rd_species %>%
mutate(species_lab = gsub("_", " ", species),
extreme_species = if_else(MDS1 > q1[2] | MDS1 < q1[1] |
MDS2 > q2[2] | MDS2 < q2[1],
"yes", "no"))
ggplot(rd_species_extreme,
aes(x = MDS1, y = MDS2, colour = group)) +
geom_point(alpha = 0.8, size = 4) +
stat_ellipse(level = 0.8, size = 1, show.legend = F) +
geom_text(data = filter(rd_species_extreme,
extreme_species == "yes"),
aes(label = species_lab), nudge_y = -0.1, show.legend = F) +
labs(x = "NMDS1", y = "NMDS2", colour = "Plant\ntype") +
lims(x = c(-1.2,1.7)) +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank())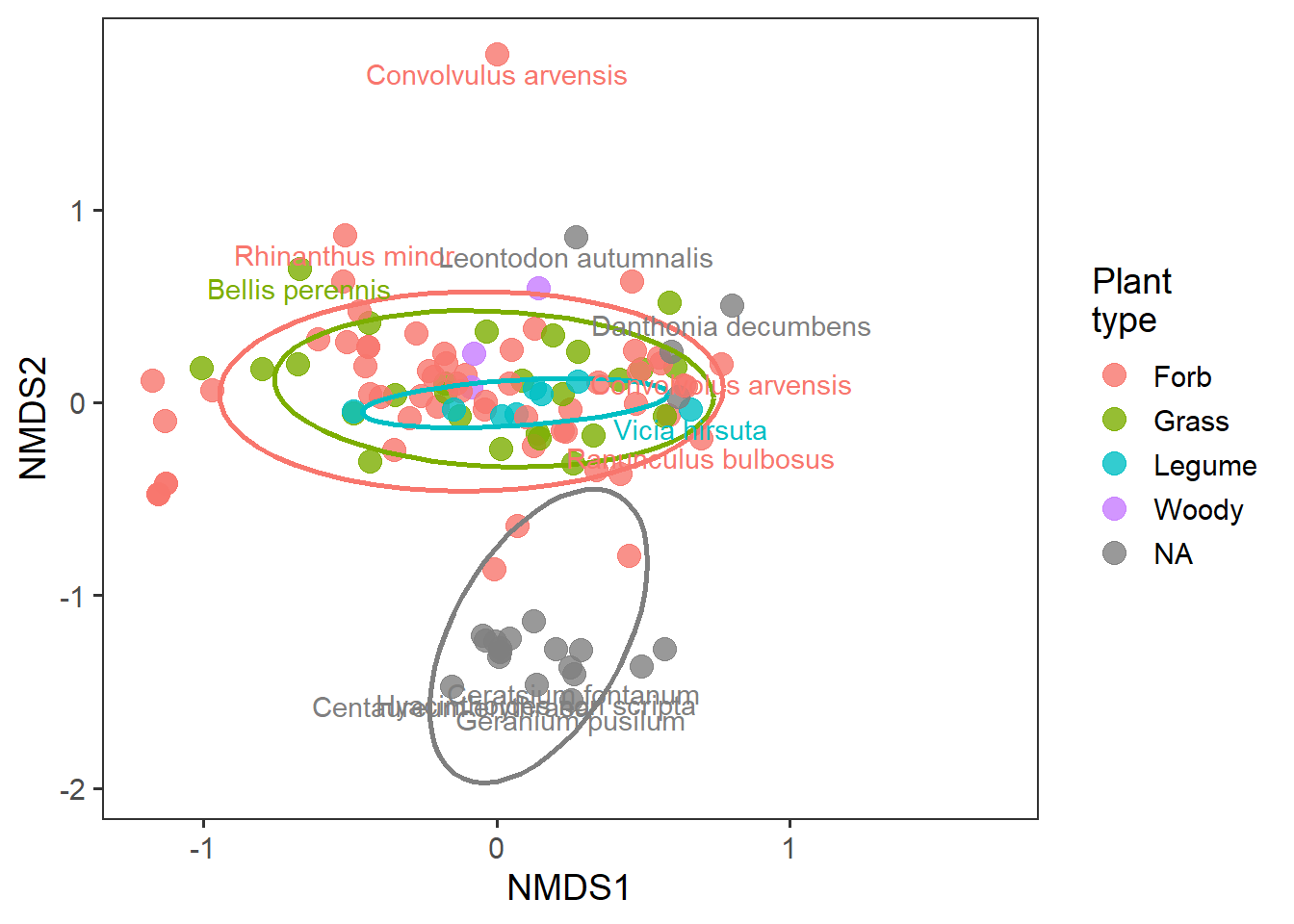
Functional group community structure
Now, rather than focusing on the whole community, we will dig down in to each functional group itself and look at the specific structure of that functional group.
# data frame of the community matrices
graminoid_community_df <- percent_cover %>%
mutate(sample = paste(month, year, block, treatment, group, sep = "_")) %>%
filter(species_level == "Yes" & group == "Grass") %>%
dplyr::select(sample, species, percent_cover) %>%
pivot_wider(id_cols = sample, names_from = species, values_from = percent_cover,
values_fill = 0)
forb_community_df <- percent_cover %>%
mutate(sample = paste(month, year, block, treatment, group, sep = "_")) %>%
filter(species_level == "Yes" & group == "Forb") %>%
dplyr::select(sample, species, percent_cover) %>%
pivot_wider(id_cols = sample, names_from = species, values_from = percent_cover,
values_fill = 0)
legume_community_df <- percent_cover %>%
mutate(sample = paste(month, year, block, treatment, group, sep = "_")) %>%
filter(species_level == "Yes" & group == "Legume") %>%
dplyr::select(sample, species, percent_cover) %>%
pivot_wider(id_cols = sample, names_from = species, values_from = percent_cover,
values_fill = 0)
# convert to matrices
graminoid_community_mat <- as.matrix(graminoid_community_df[,2:ncol(graminoid_community_df)])
rownames(graminoid_community_mat) <- graminoid_community_df$sample
forb_community_mat <- as.matrix(forb_community_df[,2:ncol(forb_community_df)])
rownames(forb_community_mat) <- forb_community_df$sample
legume_community_mat <- as.matrix(legume_community_df[,2:ncol(legume_community_df)])
rownames(legume_community_mat) <- legume_community_df$sample
# the NMDS analyses
graminoid_NMDS <- metaMDS(graminoid_community_mat, k = 3, trymax = 500)
forb_NMDS <- metaMDS(forb_community_mat, k = 3, trymax = 500)
legume_NMDS <- metaMDS(legume_community_mat, k = 3, trymax = 500)
# scores as a nice data frame
graminoid_scores <- as.data.frame(graminoid_NMDS$points) %>%
mutate(plot = rownames(.)) %>%
mutate(month = as.character(map(strsplit(plot, "_"), 1)),
year = as.numeric(map(strsplit(plot, "_"), 2)),
block = as.character(map(strsplit(plot, "_"), 3)),
treatment = as.character(map(strsplit(plot, "_"), 4)),
group = "Graminoid")
forb_scores <- as.data.frame(forb_NMDS$points) %>%
mutate(plot = rownames(.)) %>%
mutate(month = as.character(map(strsplit(plot, "_"), 1)),
year = as.numeric(map(strsplit(plot, "_"), 2)),
block = as.character(map(strsplit(plot, "_"), 3)),
treatment = as.character(map(strsplit(plot, "_"), 4)),
group = "Forb")
legume_scores <- as.data.frame(legume_NMDS$points) %>%
mutate(plot = rownames(.)) %>%
mutate(month = as.character(map(strsplit(plot, "_"), 1)),
year = as.numeric(map(strsplit(plot, "_"), 2)),
block = as.character(map(strsplit(plot, "_"), 3)),
treatment = as.character(map(strsplit(plot, "_"), 4)),
group = "Legume")
bind_rows(graminoid_scores, forb_scores, legume_scores) %>%
ggplot(aes(x = MDS1, y = MDS2, colour = treatment)) +
geom_point() +
facet_wrap(~ group) +
stat_ellipse(level = 0.8, size = 1, show.legend = F) +
scale_colour_manual(values = raindrop_colours$colour) +
labs(x = "NMDS1", y = "NMDS2", colour = "Rainfall\ntreatment") +
theme_bw(base_size = 14) +
theme(panel.grid = element_blank(),
strip.background = element_blank())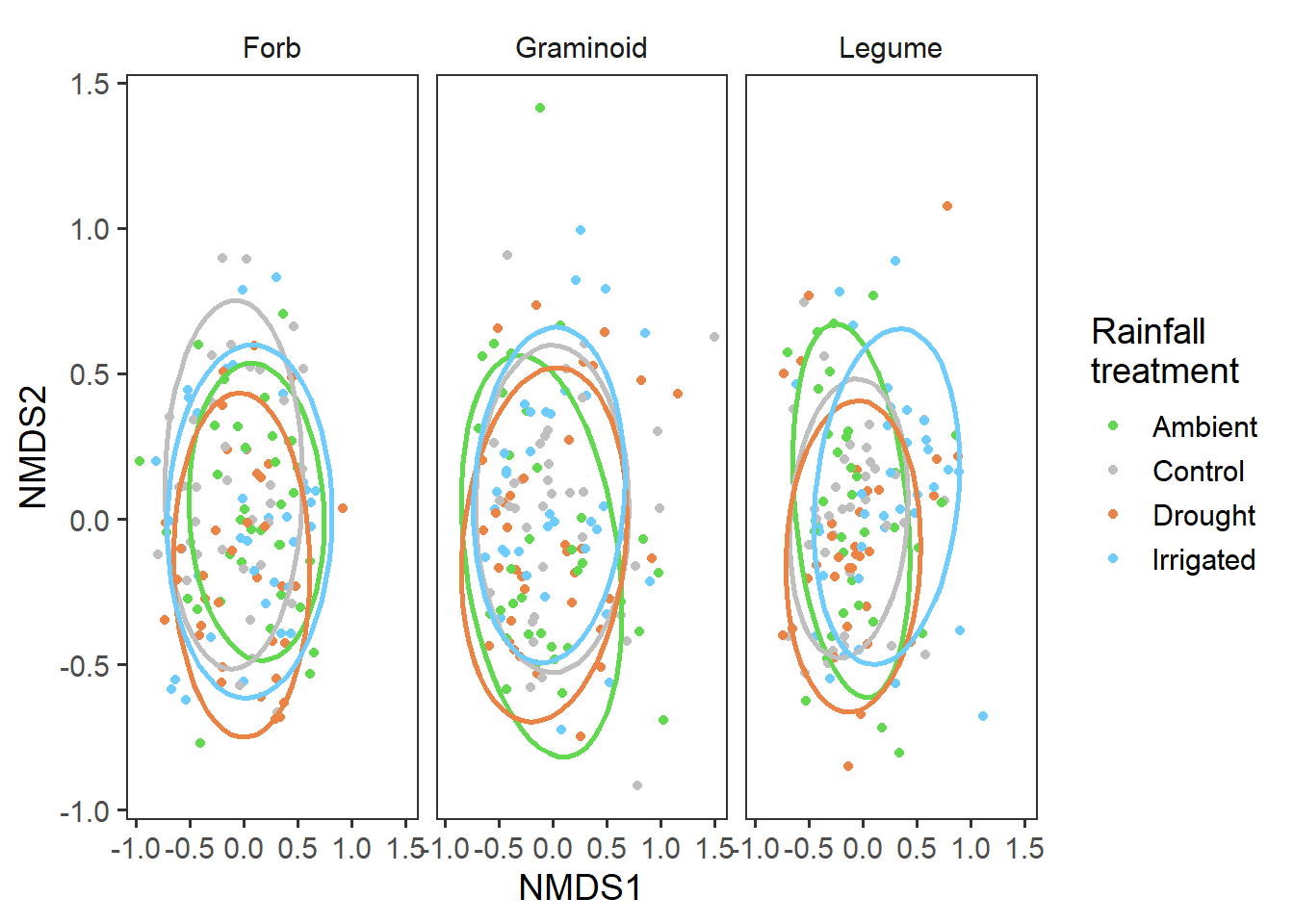
sessionInfo()R version 4.0.5 (2021-03-31)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19042)
Matrix products: default
locale:
[1] LC_COLLATE=English_United Kingdom.1252
[2] LC_CTYPE=English_United Kingdom.1252
[3] LC_MONETARY=English_United Kingdom.1252
[4] LC_NUMERIC=C
[5] LC_TIME=English_United Kingdom.1252
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] vegan_2.5-7 lattice_0.20-41 permute_0.9-5 patchwork_1.1.1
[5] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.5 purrr_0.3.4
[9] readr_1.4.0 tidyr_1.1.3 tibble_3.1.0 ggplot2_3.3.5
[13] tidyverse_1.3.1 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] httr_1.4.2 sass_0.4.0 jsonlite_1.7.2 viridisLite_0.3.0
[5] splines_4.0.5 modelr_0.1.8 bslib_0.2.5.1 assertthat_0.2.1
[9] highr_0.9 cellranger_1.1.0 yaml_2.2.1 pillar_1.6.2
[13] backports_1.2.1 glue_1.4.2 digest_0.6.27 RColorBrewer_1.1-2
[17] promises_1.2.0.1 rvest_1.0.1 colorspace_2.0-0 htmltools_0.5.1.1
[21] httpuv_1.6.1 Matrix_1.3-2 pkgconfig_2.0.3 broom_0.7.9
[25] haven_2.3.1 scales_1.1.1 whisker_0.4 later_1.2.0
[29] git2r_0.28.0 mgcv_1.8-34 generics_0.1.0 farver_2.1.0
[33] ellipsis_0.3.2 withr_2.4.2 cli_3.0.1 magrittr_2.0.1
[37] crayon_1.4.1 readxl_1.3.1 evaluate_0.14 fs_1.5.0
[41] fansi_0.4.2 nlme_3.1-152 MASS_7.3-53.1 xml2_1.3.2
[45] tools_4.0.5 hms_1.1.0 lifecycle_1.0.0 munsell_0.5.0
[49] reprex_2.0.1 cluster_2.1.1 compiler_4.0.5 jquerylib_0.1.4
[53] rlang_0.4.11 grid_4.0.5 rstudioapi_0.13 labeling_0.4.2
[57] rmarkdown_2.10 gtable_0.3.0 DBI_1.1.1 R6_2.5.0
[61] lubridate_1.7.10 knitr_1.33 utf8_1.2.1 rprojroot_2.0.2
[65] stringi_1.5.3 parallel_4.0.5 Rcpp_1.0.7 vctrs_0.3.8
[69] dbplyr_2.1.1 tidyselect_1.1.1 xfun_0.29